 达比加群酯合成路线综述.docx
达比加群酯合成路线综述.docx
- 文档编号:9497697
- 上传时间:2023-02-05
- 格式:DOCX
- 页数:18
- 大小:386.74KB
达比加群酯合成路线综述.docx
《达比加群酯合成路线综述.docx》由会员分享,可在线阅读,更多相关《达比加群酯合成路线综述.docx(18页珍藏版)》请在冰豆网上搜索。
达比加群酯合成路线综述
达比加群酯合成路线综述
一、合成路线
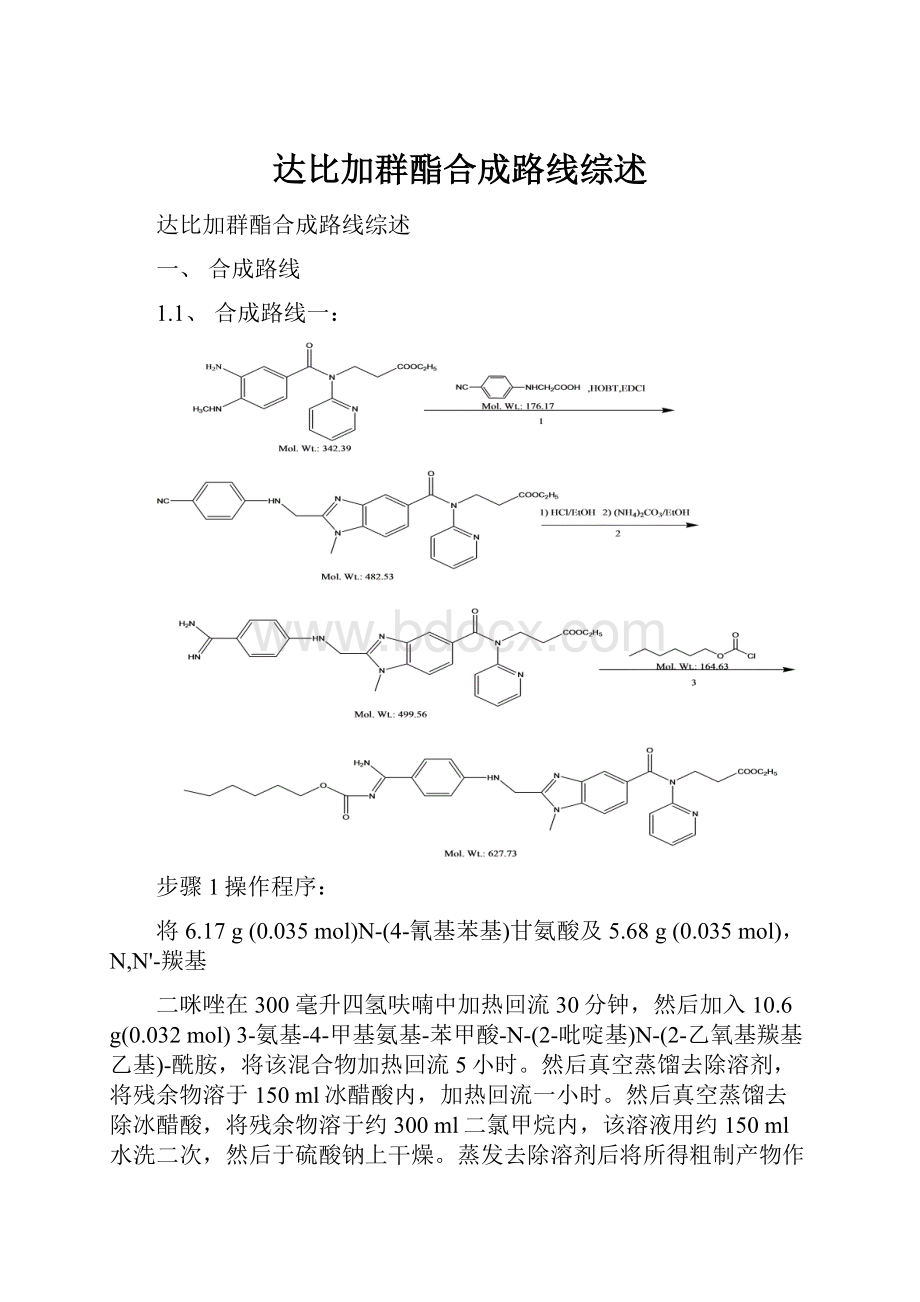
1.1、合成路线一:
步骤1操作程序:
将6.17g(0.035mol)N-(4-氰基苯基)甘氨酸及5.68g(0.035mol),N,N'-羰基
二咪唑在300毫升四氢呋喃中加热回流30分钟,然后加入10.6g(0.032mol)3-氨基-4-甲基氨基-苯甲酸-N-(2-吡啶基)N-(2-乙氧基羰基乙基)-酰胺,将该混合物加热回流5小时。
然后真空蒸馏去除溶剂,将残余物溶于150ml冰醋酸内,加热回流一小时。
然后真空蒸馏去除冰醋酸,将残余物溶于约300ml二氯甲烷内,该溶液用约150ml水洗二次,然后于硫酸钠上干燥。
蒸发去除溶剂后将所得粗制产物作柱色层纯化(800g硅胶;洗脱剂:
二氯甲烷及1-2%乙醇)。
产量8.5g,收率57%,Rf值0.51(二氯甲烷:
乙醇=19:
1)。
步骤2操作程序:
将1.2g(2.49mmol)1-甲基-2-[N-(4-氰基苯基)-氨基甲基]-苯并咪唑-5-基-羧酸-N-(2-吡啶基)-N-(2-乙氧基羟基乙基)-酰胺在100毫升饱和盐酸的乙醇溶液中在室温搅拌6小时.将该混合物在真空蒸发至干,残余物溶在100ml乙醇中与2.5g(26mmol-)碳酸铵混合,在室温下搅拌过夜.经蒸馏去除溶剂后,将所得粗制产物进行柱色层纯化(100g硅胶;洗脱剂;二氯甲烷/乙醇=4:
1)。
将洗脱液浓缩后得所需化合物,为白色固体。
产量1.10g,收率83%,Rf值0.18(二氯甲烷/乙醇=4:
1)。
步骤3操作程序:
将l.lg(2.06mmol)1-甲基-2-[N-(4-脒基苯基)-氨基甲基]-苯并咪唑-5-基-羧酸-N-(2-吡啶基)-N-(2-乙氧基羟基乙基)-酰胺盐酸盐溶于由40ml四氢呋喃和10ml水构成的混合物内,然后加入570mg(4.12mmol)碳酸钾和362mg(2.2mmol)氯甲酸正己酯,于室温搅拌二小时,然后浓缩蒸去溶剂,残余物与约50毫升饱和盐水溶液混合,所得溶液用每次用20ml二氯甲烷萃取三次.将萃取液在硫酸钠上干燥,蒸馏得粗产物进行柱色层纯化(100克硅胶;二氯甲烷+5%乙醇)。
产量0.66g,收率51%,Rf值0.53(二氯甲烷/甲醇=9:
1)。
1.2、合成路线二:
步骤1操作程序:
将2.0g(6.5mmol)3-甲基-2-[2-(4-氰基苯基)乙基]-咪唑并[4,5-b]吡啶-6-羧酸在100ml二氯甲烷中的溶液和20ml氯化亚砜混合,回流2小时.待蒸馏掉液体成分后,将粗制产物溶于二氯甲烷中二次,每次都蒸馏掉溶剂。
将这样制得酰基氯(2g)悬浮于l00ml四氢呋喃中,与1.2g(6.5mmol)N-2吡啶-B-丙氨酸乙酯混合.然后用5分钟滴加0.73g(7.2mmol)三乙胺,揽拌半小时后,真空蒸馏掉溶剂,将残余物溶于乙酸乙醋中,有机相用水洗涤,用硫酸钠干燥.浓缩掉溶剂后过柱(硅胶;二氯甲烷至二氯甲烷/乙醇=49:
1)后,分离出所需产物,为棕色油体。
1.9g,收率65%,Rf值:
0.44(乙酸乙酯/乙醇/氨水=90:
10:
1)。
步骤2操作程序:
将1.8g(3.7mmol)1-甲基-2-[N-(4-氰基苯基)-氨基甲基]-苯并咪唑-5-基-羧酸-N-(2-吡啶基)-N-(2-乙氧基羟基乙基)-酰胺加入到100ml氯化氢饱和乙醇溶液中搅拌16小时,先于0℃它搅拌,再于室温搅拌直至以TLC点板测不出有起始物料。
蒸馏掉溶剂,将油状产物溶于50ml无水乙醇中,加入3.6g(37mmol)碳酸铵,经4小时后,真空蒸馏掉溶剂,所得的粗品过柱(硅胶;梯度;二氯甲烷/乙醇19:
1至4:
1),产量1.6g,收率80%,Rf值:
0.3(乙酸乙酯/乙醇/氨水=90:
5:
5
步骤3操作程序:
将l.lg(2.06mmol)1-甲基-2-[N-(4-脒基苯基)-氨基甲基]-苯并咪唑-5-基-羧酸-N-(2-吡啶基)-N-(2-乙氧基羟基乙基)-酰胺盐酸盐溶于由40ml四氢呋喃和10ml水构成的混合物内,然后加入570mg(4.12mmol)碳酸钾和362mg(2.2mmol)氯甲酸正己酯,于室温搅拌二小时,然后浓缩蒸去溶剂,残余物与约50毫升饱和盐水溶液混合,所得溶液用每次用20ml二氯甲烷萃取三次.将萃取液在硫酸钠上干燥,蒸馏得粗产物进行柱色层纯化(100克硅胶;二氯甲烷+5%乙醇)。
产量0.66g,收率51%,Rf值0.53(二氯甲烷/甲醇=9:
1)。
1.3、合成路线三:
步骤1操作程序1:
将11.35g(70mmol)1,1'-羰基二咪唑悬浮于100mlTHF中且加热至50℃。
分批添加14.23g(60.5mmol)2-[4-(1,2,4-噁二唑-5-酮-3-基)-苯氨基]-乙酸。
将17.1g(50mmol)3-氨基-4-甲基氨基-苯甲酸-N-(2-吡啶基)N-(2-乙氧基羰基乙基)-酰胺加入到37ml四氢呋喃中,并在50℃加热下溶解。
约90min之后,将2-[4-(1,2,4-噁二唑-5-酮-3-基)-苯氨基]-乙酸悬浮液计量添加至3-氨基-4-甲基氨基-苯甲酸-N-(2-吡啶基)N-(2-乙氧基羰基乙基)-酰胺溶液中,且以20ml四氢呋喃冲洗。
将该反应混合物搅拌约18h,且接着在添加100ml乙酸后加热回流,以使四氢呋喃蒸馏掉。
约lh后,添加400ml水且搅拌该混合物。
将该溶液冷却,将所沉淀的粉红色固体物质滤出且以20ml水分2次洗涤并于真空下在最大50℃下干燥。
经分离的物质为(3)的二乙酸盐。
产量24.8g(收率75%);熔点:
167℃,纯度>95%HPLC峰面积。
步骤1操作程序2:
将34.2g(O.lmol)3-氨基-4-甲基氨基-苯甲酸-N-(2-吡啶基)N-(2-乙氧基羰基乙基)-酰胺、27.5g(O.12mol)2-[4-(1,2,4-噁二唑-5-酮-3-基)-苯氨基]-乙酸及30.3g(O.23mol)二异丙基乙胺置于170ml四氢呋喃中且冷却至稍低于周围温度。
接着计量添加85g(0.13mol)丙烷磷酸酐(乙酸乙酯中约50%的溶液)。
将该混合物再搅拌90分钟且接着将溶剂蒸馏掉。
接近终点时添加73.5g乙酸且将该混合物加热至90℃的内部温度。
接着添加400ml乙醇或优选400ml乙醇/水(约85:
15)且将该混合物热过滤。
将该溶液冷却,将沉淀的固体物质滤出且以50ml乙醇分2次洗涤及于真空下在最大50℃下干燥。
经分离的物质为(3)的二乙酸盐。
产量56g(收率75%);熔点:
167℃,纯度>95%HPLC峰面积。
步骤1操作程序3:
于0℃下,将96g(0.41mol)2-[4-(1,2,4-噁二唑-5-酮-3-基)-苯氨基]-乙酸悬浮于250mlN-甲基吡咯烷酮及550ml四氢呋喃中。
继而将该稀的悬浮液与48g(0.4mol)三甲基乙酰基氯及52g(0.4mol)二异丙基乙胺混合且搅拌30分钟。
接着添加溶解于800ml乙酸中的125g(0.36mol)3-氨基-4-甲基氨基-苯甲酸-N-(2-吡啶基)N-(2-乙氧基羰基乙基)-酰胺,且将该反应混合物加热回流3h。
在轻微真空下将四氢呋喃蒸馏掉且于温热时计量添加1600ml水。
将该固体于5℃下分离,以550ml水洗涤并于循环空气干燥器中在最大50℃下干燥过夜。
步骤2操作程序1:
将37.3g(56.4mmol)1-甲基-2-[N-[4-(1,2,4-噁二唑-5-酮-3-基)-苯基]-氨基-甲基]-苯并咪唑-5-基羧酸-N-(2-吡啶基)-N-(2-乙氧基羰基乙基)-酰胺溶解于900ml乙醇中,且在添加10ml乙酸后,于室温下及2巴氢气下以4g经水潮湿的10%Pd/C氢化。
将催化剂滤出且将溶解于180ml乙醇中的17g(89.4mmol)对甲苯磺酸添加至该滤液。
将的1-甲基-2-[N-(4-脒基苯基)-氨基甲基]-苯并咪唑-5-基-羧酸-N-(2-吡啶基)-N-(2-乙氧基羟基乙基)-酰胺的甲苯磺酸盐沉淀出,滤出且再次以150ml乙醇分多次洗涤。
获得潮湿物质,将其于35℃真空下干燥。
产量34.5g,浅米色物质(收率91.3%),熔点187℃,纯度:
>98%HPLC峰面积。
步骤2操作程序2:
将37.3g(56.4mmol)溶解于1-甲基-2-[N-[4-(1,2,4-噁二唑-5-酮-3-基)-苯基]-氨基-甲基]-苯并咪唑-5-基羧酸-N-(2-吡啶基)-N-(2-乙氧基羰基乙基)-酰胺400ml乙醇/水(90:
10)中,且室温于及2巴氢气下,以4g经水潮湿的10%Pd/C氢化。
将催化剂过滤且将11.5g(60.6mmol)对甲苯磺酸添加至该滤液。
经蒸发使(4)的甲苯磺酸盐沉淀出。
将该悬浮液冷却,将该物质滤出且以150ml乙醇/水分多次洗涤。
获得潮湿物质,将其于35℃真空下干燥。
产量33.7g,浅米色物质(收率89%),熔点187℃,纯度:
>98%HPLC峰面积。
步骤2操作程序3:
室温下,将30.0g(45.3mmol)1-甲基-2-[N-[4-(1,2,4-噁二唑-5-酮-3-基)-苯基]-氨基-甲基]-苯并咪唑-5-基羧酸-N-(2-吡啶基)-N-(2-乙氧基羰基乙基)-酰胺溶解于90mlTHF/水(1:
1)中,与4g经水潮湿的10%Pd/C混合且于4巴氢气下,60℃下氢化。
将催化剂滤出,再以大约40ml的THF/水(l:
1)洗涤且将滤液无需处理用于下一步骤,或如上文所述通过添加溶解于100ml水中的13.6g(72mmol)对甲苯磺酸进行分离并冷却。
步骤3操作程序1:
在有34g(246mmol)碳酸钾存在下,在约15℃的温度下,将溶解于437ml丙酮及273ml水中的55g(81.9mmol)1-甲基-2-[N-(4-脒基苯基)-氨基甲基]-苯并咪唑-5-基-羧酸-N-(2-吡啶基)-N-(2-乙氧基羟基乙基)-酰胺与16.4g(99.6mmol)氯甲酸己酯混合。
反应结束后,将析出的产物滤出且以丙酮/水洗涤。
必要时,可将其在加热下再次溶解在约270ml丙酮中,且接着过滤。
过滤后,通过添加220ml水
使该物质再次结晶。
将分离的物质于45℃真空下干燥。
产量42-48g,收率82-94%。
步骤3操作程序2:
在有34g(246mmol)碳酸钾存在下,在约15℃的温度下,将溶解于437ml丙酮及273ml水中的55g(81.9mmol)1-甲基-2-[N-(4-脒基苯基)-氨基甲基]-苯并咪唑-5-基-羧酸-N-(2-吡啶基)-N-(2-乙氧基羟基乙基)-酰胺与16.4g(99.6mmol)氯甲酸己酯混合。
反应结束后,将该悬浮液加热至约50℃。
分离有机相且由440ml乙酸乙酯代替丙酮。
将接着分离的水相丢弃且将有机相以稀释的碳酸钾溶液多次洗涤并最后以水洗涤。
将产物冷却析晶,分离及以乙酸乙酯洗涤。
45℃真空下干燥。
产量42-48g,收率82-94%。
步骤4操作程序:
将l00g(O.16mol)1-甲基-2-[N-[4-(N-正己氧基羰基甲脒基)苯基]-氨基甲基]-苯并咪唑-5-基-羧酸-N-(2-吡啶基)-N-(2-乙氧基羟基乙基)-酰胺在加热下溶解于890ml丙酮中且与15g(O.16mol)甲磺酸于200ml丙酮中的溶液混合。
将该溶液过滤,加77ml丙酮后冷却至约20℃。
将析出的产物分离且以丙酮同再次洗涤。
50℃以下真空下干燥。
产量103-113,收率90-98%。
1.4、合成路线四:
步骤1操作程序:
Calciumchloridedihydrate(12.5g)wasaddedtoamixtureofl-methyl-2-[N-(4-cyanophenyl)aminomethyl]benzimidazol-5-ylcarboxylicacid-N-(2-pyridyl)-N-(2-ethoxycarbonylethyl)amidecompoundofformula-14(50g)andethanol(750ml)andstirredfor20minutes.Thereactionmixturewascooledto0-5℃.andHClgaswaspassedintothereactionmixtureoveraperiodof5hoursatatemperature
below10℃.Thetemperatureofthereactionmixturewasraisedto25-30.andstirredfor8hoursatthesametemperature.Aftercompletionofthereaction,thesolventwasexpelledoutunderN2pressure.Thereactionmixturewascooledto0-5℃.andslowlyaddedammoniumformate(150g).Thereactionmixturewasstirredfor30minutesandammoniumcarbonate(300g)wasadded.Thetemperatureofthereactionmixturewasraisedto25-35℃.andstirredfor10hours.Aftercompletionofthereaction,thereactionmixturewasfilteredandthefiltratewasdistilledunderreducedpressure.Asolutionof10%ethanolinethylacetatewasaddedtothereactionmixtureandstirredfor3hourstoobtainasolid.Filteredtheobtainedsolid,washedwithethylacetateandthendriedtogetthetitlecompound.Yield:
45g.
Amixtureof1-methyl-2-[N-[4-amidinophenyl]aminomethyl]benzimidazol-5-yl-carboxylicacid-N-(2-pyridyl)-N-(2-ethoxycarbonylethyl)amidecompoundofformula-5(100g)andethanol(1200ml)washeatedto50-60℃.Asolutionofoxalicacid(25.25g)inethanol(1500ml)wasaddedtotheabovereactionmixtureat50-60℃.andstirredfor45minutes.Thereactionmixturewascooledto25-35℃.andstirredfor6hoursat25-35℃.Filteredthesolid,washedwithethanolandthendriedtogetthetitlecompound.
室温下,在50g1-甲基-2-[N-(4-氰基苯基)-氨基甲基]-苯并咪唑-5-基-羧酸-N-(2-吡啶基)-N-(2-乙氧基羟基乙基)-酰胺中加入乙醇750ml,加入CaCl212.5g,搅拌20min后降温至0-5℃。
控温10℃以下,通入HCl气体搅拌反应5h。
升温至25-30℃,搅拌反应8h。
反应完成后,通入N2吹干乙醇。
降温至0-5℃,缓慢加入甲酸铵150g。
在30min内加入碳酸铵300g。
升温至25-35℃,搅拌反应10h。
反应完成后,将反应混合物过滤,将滤液在减压下蒸馏。
10%乙醇的乙酸乙酯溶液加入到反应混合物中,并搅拌3小时,得到固体。
过滤得到的固体,用乙酸乙酯洗涤,然后干燥,得到目标化合物。
产量:
45g。
在50g1-甲基-2-[N-(4-脒基苯基)-氨基甲基]-苯并咪唑-5-基-羧酸-N-(2-吡啶基)-N-(2-乙氧基羟基乙基)-酰胺中加入1200乙醇,升温至50-60℃,滴加25.25g草酸加1500乙醇溶液,滴加完毕后搅拌45min。
降温至25-35℃搅拌6h。
过滤,滤饼用乙醇洗涤,真空干燥的产品。
步骤2操作程序:
Asolutionofn-hexanol(25.95g)indichloromethane(400ml)wasslowlyaddedtosolutionofN,N-carbonyldiimidazole(48.08g)indichloromethane(100ml)andstirredfor21hhourat25-35℃.Waterwasaddedtothereactionmixture.Boththedichloromethanelayerandaqueouslayerwereseparatedandthedichloromethanelayerwasdistilledunderreducedpressuretoprovidethetitlecompound.Yield:
60g
向25.95g正己醇400ml二氯甲烷的溶液中缓慢滴加48.08gN,N羰基二咪唑+100ml二氯甲烷溶液,控温25-35℃,搅拌反应21h。
反应完成后,加入纯水,分离有机相,有机相减压浓缩二氯甲烷得产品60g。
步骤3操作程序:
1-methyl-2-[N-[4-amidinophenyl]aminomethyl]benzimidazol-5-yl-carboxylicacid-N-(2-pyridyl)-N-(2-ethoxycarbonylethyl)amideoxalatecompoundofformula-6a(100g)wasaddedtoacetonitrile(1200ml)andwater(800ml)at25-35℃.andthencooledto12-18℃.Potassiumcarbonate(117g)wasaddedtothereactionmixtureandstirredfor15minutesat12-18℃.Asolutionofhexyl1H-imidazole-l-carboxylatecompoundofformula-4(60g)inacetonitrile(150ml)wasslowlyaddedtothereactionmixtureoveraperiodof25minutesat12-18℃.andstirredfor14hoursat15-20℃.Aftercompletionofthereaction,waterwasaddedtothereactionmixtureandstirredfor30minutes.Filteredthesolid,washedwithacetonitrilefollowedbyaqueousacetonitrileandthendriedtogettitlecompound.Dichloromethanewasaddedtotheobtainedcompoundandstirredfor15minutes.Waterwasaddedtothereactionmixtureandstirredfor20minutesat25-35℃.Boththeorganicandaqueouslayerswereseparated,andthedichloromethanelayerwaswashedwithwaterfollowedbysodiumchlorideandthendistilledoffcompletelyunderreducedpressure.Acetone(600ml)wasaddedtotheobtainedresidueandstirredfor45minutesat25-35℃.toobtainaclearsolution.Water(500ml)wasaddedtotheobtainedsolutionandstirredfor45minutesat25-35℃.togetthesolid.Filteredthesolid,washedwithwaterandfinallywithmethyltertiarybutyletherandthendriedtogetthepuretitlecompound.Furthertheobtainedsolid,recrystallizedfromethylacetateandethanol.
控温25-35℃,在1-甲基-2-[N-(4-脒基苯基)-氨基甲基]-苯并咪唑-5-基-羧酸-N-(2-吡啶基)-N-(2-乙氧基羟基乙基)-酰胺草酸盐中加入乙腈1200ml,纯水800ml。
降温至12-18℃,加入碳酸钾117g,搅拌15min。
控温12-18℃,在25min内缓慢加入60g1,1'-羰基-正己氧基-咪唑,150ml乙腈溶液,搅拌反应14h。
反应完成后,加入纯水析晶搅拌30min。
过滤,滤饼分别用乙腈和乙腈水溶液洗涤,干燥。
二氯甲烷溶解上步所得化合物,搅拌15min,加入纯水搅拌,静置,分层。
有机相用饱和氯化钠溶液洗涤,真空浓缩干溶剂。
加入丙酮600ml,控温25-35℃搅拌45min至溶液溶清,加入纯水500ml,控温25-35℃搅拌45min,过滤,分别用纯水,甲基叔丁基醚洗涤滤饼,真空干燥得产品。
步骤4操作程序:
AsolutionofDabigatranetexilatecompoundofformula-l(100g)inethylacetate(600ml)washeatedto40℃.andstirredfor45minutesat40℃Filteredthereactionmixturethroughhyflowbedandcooledto25-30°C.Ethanol(60ml)wasaddedtothefiltrateat25-35℃.Asolutionofmethanesulfonicacid(15g)inethylacetate(1000ml)wasslowlyaddedtothe


- 配套讲稿:
如PPT文件的首页显示word图标,表示该PPT已包含配套word讲稿。双击word图标可打开word文档。
- 特殊限制:
部分文档作品中含有的国旗、国徽等图片,仅作为作品整体效果示例展示,禁止商用。设计者仅对作品中独创性部分享有著作权。
- 关 键 词:
- 加群酯 合成 路线 综述
 冰豆网所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
冰豆网所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
 1212中级汽车维修工考试试题三.docx
1212中级汽车维修工考试试题三.docx
-
333教育综合.docx
-
204届毕业生基础知识考试试题 混凝土结构设计 试题.docx
-
100以内加减运算练习题.docx
-
101软件开发工程师JAVA初级考试样卷课件word版本.docx
-
CNN代码理解.docx
-
CPA审计第4章审计抽样下载版讲解.docx
-
hr培训管理系统.docx
-
318安通科科长岗位责任制.docx
-
2044施工现场环境污染的防治措施.docx
-
12371党务平台操作手册.docx
-
Catia百格线生成宏复习过程.docx
-
725kV及以上电压等级支柱瓷绝缘子运行规范.docx
-
1144甑底链板机说明书.docx
-
100个著名初等数学问题.docx
-
201X中学寒假工作计划范文.docx
-
111 生物的特征 练习 人教版七年级上册生物.docx
-
110KV变电所设计变压器翻译.docx
-
9920第二学期学校工作总结.docx
-
0911二级技能解答.docx
-
33415设计说明书正文.docx
-
311教育学基础综合大纲.docx
-
201浙江普通高校招生选考科目考试地理试题和答案解析.docx
-
C语言程序的设计实验实验指导书及答案.docx
-
272相似三角形的性质和判定.docx
-
ACCAHA不稳定型心绞痛和非ST段抬高心肌梗死治疗指南修订版摘要.docx
-
baosteel标准对照 外标含量.docx
-
M1模拟练习题.docx
-
ARM体系课程设计实验报告.docx
-
Android面试题整理.docx
-
gaoer.docx
-
CPⅢ测设方案.docx
-
临考必备考研考前准备实用手册完整版.docx
-
内分泌系统的解剖生理.docx
-
山西高考文综试题和答案新课标.docx
-
化工泵类设备常规知识.docx
-
年产5000吨连续挤压无氧银铜异型排项目可行性研究报告可行性研究报告.docx
-
美国第三次剪羊毛不成已经疯了讲解.docx
-
善良的吃人巨魔.docx
-
各国企业管理比较.docx
-
高考英语词汇大全附例句.docx
-
客服年终个人工作总结范文.docx
-
工伤事故管理制度处理办法.docx
-
六年级诵读.docx
-
尼龙材料相关资料整理.docx
-
领导班子自我评价材料.docx
-
怀柔区中考二模英语试题及答案.docx
-
尼采最经典的哲理名言16句说课讲解.docx
-
各类工作职责.docx
-
上半年安徽省综合法律知识自然资源的行政管理制度模拟试题.docx
-
宁波市文联文艺创作与人才培养规划.docx
