 MaterialStudio建模.docx
MaterialStudio建模.docx
- 文档编号:603624
- 上传时间:2022-10-11
- 格式:DOCX
- 页数:21
- 大小:1.22MB
MaterialStudio建模.docx
《MaterialStudio建模.docx》由会员分享,可在线阅读,更多相关《MaterialStudio建模.docx(21页珍藏版)》请在冰豆网上搜索。
MaterialStudio建模
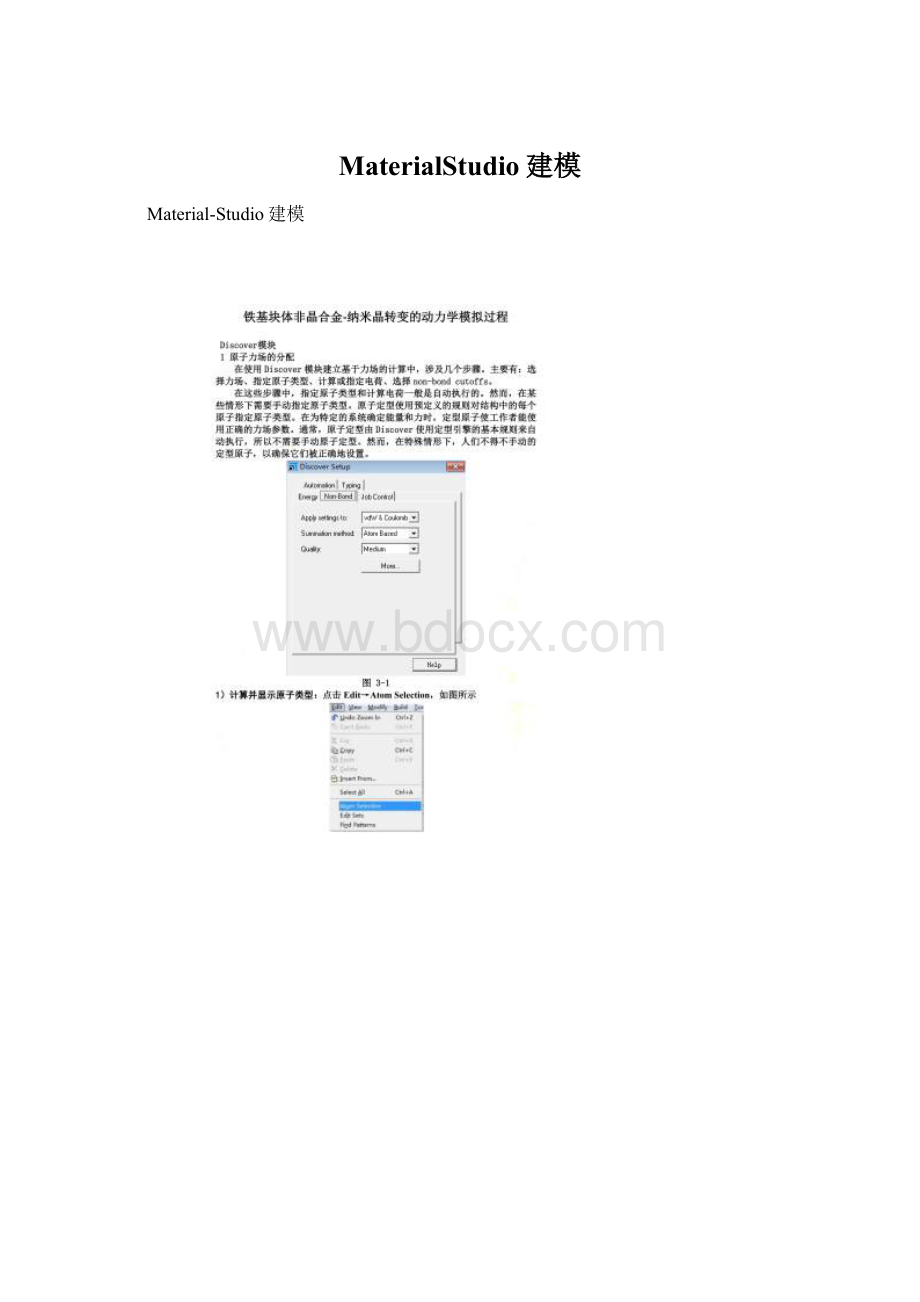
Material-Studio建模
图3-2DiscoverSetup对话框Typing选项卡
在Forcefieldtypes里选择相应原子力场,再点Assign(分配)按钮进行原子力场分配。
注意原子力场中的价态要与PropertiesProject里的原子价态(Formalcharge)一致。
2力场的选择
1)Energy
力场的选择:
力场是经典模拟计算的核心,因为它代表着结构中每种类型的原子与围绕着它的原子是如何相互作用的。
对系统中的每个原子,力场类型都被指定了,它描述了原子的局部环境。
力场包括描述属性的不同的信息,如平衡键长度和力场类型对之间的电子相互作用。
常见力场有COMPASS、CVFF和PCFF。
Select下拉菜单中有三个选项:
①COMPASS力场:
COMPASS力场是第一个把以往分别处理的有机分子体系的力场与无机分子体系的力场统一的分子力场。
COMPASS力场能够模拟小分子与高分子,一些金属离子、金属氧化物与金属。
在处理有机与无机体系时,采用分类别处理的方式,不同的体系采用不同的模型,即使对于两类体系的混合,仍然能够采用合理的模型描述。
②CVFF力场:
CVFF力场全名为一致性价力场(consistantvalenceforcefield),最初以生化分子为主,适应于计算氨基酸、水及含各种官能团的分子体系。
其后,经过不断的强化,CVFF力场可适用于计算多肽、蛋白质与大量的有机分子。
此力场以计算系统的结构与结合能最为准确,亦可提供合理的构型能与振动频率。
③PCFF力场:
PCFF为一致性力场,增加一些金属元素的力参数,可以模拟含有相应原子的分子体系,其参数的确定除大量的实验数据外,还需要大量的量子力学计算结果。
3非键的设置:
非键作用力包括范德华力和库伦力。
这里将两者都选上,为的是后期做minimizer优化原子位置时精确度更高,因为考虑了作用力因素多,即两者都考虑了。
Summationmethod(模拟方法):
1AtomBased
atombased基于原子的总量,包括一个原子的截断距离,一个原子的缓冲宽度距离;为直接计算法,即直接计算原子对之间的非键相互作用,当原子对超出一定距离(截断半径cutoffdistance)时,即认为原子对之间相互作用为零(注:
cutoffdistance指范德瓦尔斯作用力和库仑力的范围,比如:
设定截断半径为5,则表示已分子或原子中心为圆心,以5为半径作圆,半径以外的作用力都不考虑)。
此方法计算量较小,但是可能导致能量和其导数的不连续性。
当原子对间距离在CutOff半径附近变化时,由于前一步考虑了原子对之间的相互作用,而后一步不考虑,由此会导致能量发生跳跃。
当然,对于较小的体系,则可以设置足够大的Cutoff半径来保证所有的相互作用都被考虑进来。
2GroupBased
groupbased基于电子群的,总量中包括一个原子的截断距离,一个原子的缓冲宽度距离;大多数的分子力场都包括了每个原子之间点电荷的库仑相互作用。
甚至在电中性的物种中也存在点电荷,例如水分子。
点电荷实际上反映了分子中不同原子的电负性。
在模拟中,点电荷一般是通过电荷平衡法(chargeequilibrium)评价或者力场定义的电荷来分配的。
当评价点电荷时,一定要小心不要在使用Cutoff技术时引入错误的单极项。
要了解到这一点,可以参看如下事实:
两个单极,当只有1e.u.电荷时,在10A的位置上其相互作用大约为33Kcal;而对于由单位单极分离1A所形成的两个偶极,相同距离其相互作用能不超过0.3Kcal/mol。
很明显,忽略单极-单极相互作用会导致错误的结果,而忽略偶极-偶极相互作用则是适度的近似。
然而,如果单极相互作用处理不清的话,仍然会出问题。
当non-bondCutoff使用基于原子-原子基组时,就可能发生,会人为将偶极劈裂为两个“假”的单极(当一个偶极原子在Cutoff内,另一个在其外)。
这就不是忽略了相对较小的偶极-偶极相互作用,而是人为引入了作用较大的单极-单极相互作用。
为了避免这种人为现象,MaterialsStudio引入了在ChargeGroups之上的Cutoff。
一个“ChargeGroup”是一个小的原子基团,其原子彼此接近,净电荷为0或者接近于0。
在实际应用中,ChargeGroup一般是常见的化学官能团,例如羰基、甲基或者羧酸基团的净电荷接近于中性ChargeGroup。
ChargeGroup之间的距离为一个官能团中心到另一个官能团中心的距离R,Cutoff设置与AtomBased相类似。
3EwaldSummation
Ewald是在周期性系统内计算Non-bond的一种技术。
Ewald是计算长程静电相互作用能的一种算法。
Ewald加和方法比较合适于结晶固体。
原因在于无限的晶格内,Cutoff方法会产生较大的误差。
然而,此方法放也可以用于无定形固体和溶液体系。
Ewald计算量较大,为o(N^3/2),体系较大时,会占用较多的内存并花费较长的时间【《分子模拟—从算法到应用》】。
4cellmultipolecellbased只能用于基于指定数量层。
一般情况下,基于Atom适合于孤立体系,对于周期性体系计算量较小,但是准确性较差;基于Group适合于周期性和非周期性体系,计算的准确性好一些,计算量最小;Ewald适合于周期性能体系,计算最为准确,但计算量最大。
Cutoffdistance(截断距离):
指的是范德瓦尔斯作用力和库仑力的范围。
Splinewidth:
Bufferwidth:
缓冲宽度距离。
Setup其他选项保留默认设置即可。
4结构优化
在工具栏上点击Discover按钮,然后选择Minimizer。
或者从菜单栏选择Modules|Discover|Minimizer。
显示DiscoverMinimizer对话框,可以进行几何结构优化计算。
注:
优化前(Min),先查看所有原子是否都已分配力场,如果没有,可以手动添加,在PropertiesExplorer中双击Forcefieldtype,然后修改力场类型即可。
其次在Min之前,需要把晶体结构所有原子重新固定。
minimizer只是对结构进行优化,以达到能量最小化。
在作动力学(dynamics)之前最好minimizer一下,因为如果不minimizer计算收敛时间会比较长,能量波动会比较大,而且计算有可能出错。
优化方法Mathod:
最陡下降法(SteepestDescent)、共轭梯度法(ConjugateGradient)、牛顿方法(Newton)和综合法(SmartMinimizer)。
Convergencelevel:
收敛精度水平。
Maximumiteration:
最大迭代数。
Optimizecell选中的话表示优化晶胞参数和原子位置。
MSDiscover结构优化原理
分子的势能一般为键合(键长、键角、二面角、扭转角等)和非键合相互作用(静电作用、范德华作用等)能量项的加和,总势能是各类势能之和,如下式:
除了一些简单的分子以外,大多数的势能是分子中一些复杂形势的势能的组合。
势能为分子中原子坐标的函数,由原子不同的坐标所得到的势能构成势能面(PotentialEnergySurface,PES)。
势能越低,构象越稳定,在系统中出现的机率越大;反之,势能越高,构象越不稳定,在系统中出现的机率越小。
通常势能面可得到许多极小值的位置,其中对应于最低能量的点称为全局最小值(GlobalEnergyMinimum),相当于分子最稳定的构象。
由势能面求最低极小值的过程称为能量最小化(EnergyMinimum),其所对应的结构为最优化结构(OptimizedStructure),能量最小化过程,亦是结构优化的过程。
通过最小化算法进行结构优化时,应避免陷入局部最小值(localminimum),也就是避免仅得到某一构象附近的相对稳定的构象,而力求得到全局最小值,即实现全局优化。
分子力学的最小化算法能较快进行能量优化,但它的局限性在于易陷入局部势阱,求得的往往是局部最小值,而要寻求全局最小值只能采用系统搜寻法或分子动力学法。
在MaterialsStudio的Discover模块中,能量最小化算法有以下四种:
1)最陡下降法(SteepestDescent),为一经典的方法,通过迭代求导,对多变量的非线性目标函数极小化,按能量梯度相反的方向对坐标添加一位移,即能量函数的负梯度方向是目标函数最陡下降的方向,所以称为最陡下降法。
此法计算简单,速度快,但在极小值附近收敛性不够好,造成移动方向正交。
最陡下降法适用于优化的最初阶段。
2)共轭梯度法(ConjugateGradient),在求导时,目标函数下降方向不是仅选取最陡下降法所采用的能量函数的负梯度方向,而是选取两个共轭梯度方向,即前次迭代时的能量函数负梯度方向与当前迭代时的能量函数负梯度方向的线性组合。
此法收敛性较好,但对分子起始结构要求较高,因此常与最陡下降法联合使用,先用最陡下降法优化,再用共轭梯度法优化至收敛。
3)牛顿方法(Newton),以二阶导数方法求得极小值。
此法的收敛很迅速,也常与最陡下降法联合使用。
4)综合法(SmartMinimizer),该方法可以混合最陡下降法,共轭梯度法和牛顿法进行结构优化,在MS中是可选择的。
SmartMinimizer中,牛顿法可以设定最大的原子数,如果体系的原子数大于所设定的值,则计算是会自动地转为前面设定的收敛法(共轭梯度法或最陡下降法),收敛精度会改为共轭梯度法的默认收敛精度值。
点开各种方法后面的More,可设定收敛精度(Convergence),算法(Algorithm)和一维搜索(Linesearch,指每一次迭代中的精度)等。
当Job结束后,结果被返回到DiscoMin目录,最小化的结构被命名为3DAtomistic.xsd,并被保存在“3DAtomisticDiscoMin”目录。
还生成一个名为“3DAtomistic.out”的文本文档,它包含了有关计算的所有能量信息。
同时还生成“SimulationEnergies.xcd”,它显示了能量随迭代次数的变化情况。
如图所示。
本次模拟得到如图所示的结构,
5高温弛豫
打开discover下的Dynamics,如图所示
Ensemble(系综):
NVE、NVT、NPT、NPH。
Temperture:
目标温度。
Pressure:
给系统所施加的压力。
Numberofsteps:
整个动力学所运行的总步数。
Timpstep:
每一动力学步骤所花费的时间。
Dynamicstime:
Numberofsteps×Timpstep。
TrajectorySave:
Coordinates表示保存坐标;FinalStructure表示只保存最终结构;Full表示保存所有。
Frameoutputevery:
若输入5000,则表示每5000步输出一次体系构型文件。
此操作表示结构在2000K的温度下进行弛豫,此过程原子通过迁移、运动或者扩散,逐步降低原来的高内能态,向稳定的低内能态转变。
运行结束后,可以通过调用Animation观看三维动画,见图
动画工具条可以控制三维窗口中动画文件的显示。
它包含以下命令:
PlayBackwards:
倒映动画文件。
StepBackwards:
每次向后放一帧
Stop:
停止放映。
StepForwards:
每次一帧加速放映。
Play:
放映动画。
Pause:
暂停放映,再按一次


- 配套讲稿:
如PPT文件的首页显示word图标,表示该PPT已包含配套word讲稿。双击word图标可打开word文档。
- 特殊限制:
部分文档作品中含有的国旗、国徽等图片,仅作为作品整体效果示例展示,禁止商用。设计者仅对作品中独创性部分享有著作权。
- 关 键 词:
- MaterialStudio 建模
 冰豆网所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
冰豆网所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
 转基因粮食的危害资料摘编Word下载.docx
转基因粮食的危害资料摘编Word下载.docx
-
高中英语词组大全Word文档下载推荐.docx
-
卫计局年工作总结及新年工作计划Word格式.docx
-
贵州省煤矿安全管理人员安全资格证A考试概况Word格式.docx
-
系统集成项目招标文件Word文件下载.docx
-
电子商务考试题总汇打印版打印打印Word下载.docx
-
选调生考试备考言语理解与表达真题Word文档格式.docx
-
高考物理实验题专练 专练15Word文档格式.docx
-
加装奥迪A4L蓝牙电话功能Word文档下载推荐.docx
-
学年下学期好教育高三月考仿真卷A卷 语文 学生版后附详解Word文档下载推荐.docx
-
净化生产车间工程一般施工技术施工方案Word文档格式.docx
-
内蒙古呼和浩特市第六中学学年高一政治下学期期末考试试题Word下载.docx
-
证券行业客户经理电话营销技巧与实例Word文档下载推荐.docx
-
叶芝 苇间风文档格式.docx
-
最新中美贸易摩擦的原因及解决对策1论文Word文件下载.docx
-
意义的近义词Word格式文档下载.docx
-
上海市中考英语试题S.docx
-
专题12观点论证类设问.docx
-
附加安心重疾条款.docx
-
设计变更管理办法修改意见稿FINAL汇编.docx
-
毕业赠言毕业致词精选多篇.docx
-
银行新员工代表发言稿精选多篇.docx
-
北京市朝阳区届高三第一学期期末语文试题Word版含答案.docx
-
HL线切割使用说明书模板.docx
-
车工实训周记.docx
-
USBHID键盘扫描码.docx
-
Apmpoqu4调研报告.docx
-
最熟悉的陌生人作文八篇.docx
-
被动语态综合讲解.docx
-
部编版语文七上第五单元16猫同步练习试题.docx
-
软件体系结构作业2.docx
-
钢管管道安装焊接施工工艺.docx
-
《依托儿童文学培养小学高段学生良好阅读习惯的研究》结题报告Word格式.docx
-
实施方案资中县中医医院迁建工程建设项目文档格式.docx
-
青蛇Word下载.docx
-
饰品专卖店陈列培训手册全Word文档下载推荐.docx
-
英语教师工作总结Word格式.docx
-
超声波清洗机控制系统设计课程设计任务书Word文档下载推荐.docx
-
软件测试标准和测试用例汇总Word下载.docx
-
大藤峡水利枢纽建设征地长期补偿安置方式研究Word文档格式.docx
-
软件测试标准和测试用例汇总Word格式文档下载.docx
-
青蛇Word文档下载推荐.docx
-
综合布线实验Word格式.docx
-
祝寿流程规范Word下载.docx
-
意识及潜意识的本质区别Word格式.docx
-
用ghost备份还原安装版windows 10正确方法Word格式.docx
-
学年安徽省蚌埠市固镇县八年级上册期末数学试题有答案标准版Word下载.docx
-
融资商品车监管流程Word格式.docx
-
《燕子专列》教学设计三Word文档格式.docx
-
人教版小学语文三年级上册期中学习质量检测卷Word文档下载推荐.docx
-
资料Word下载.docx
