 颅内T低信号总结包括正常及病变.docx
颅内T低信号总结包括正常及病变.docx
- 文档编号:5576288
- 上传时间:2022-12-28
- 格式:DOCX
- 页数:7
- 大小:107.85KB
颅内T低信号总结包括正常及病变.docx
《颅内T低信号总结包括正常及病变.docx》由会员分享,可在线阅读,更多相关《颅内T低信号总结包括正常及病变.docx(7页珍藏版)》请在冰豆网上搜索。
颅内T低信号总结包括正常及病变
T1、T2低信号——影像2011-07-1212:
24
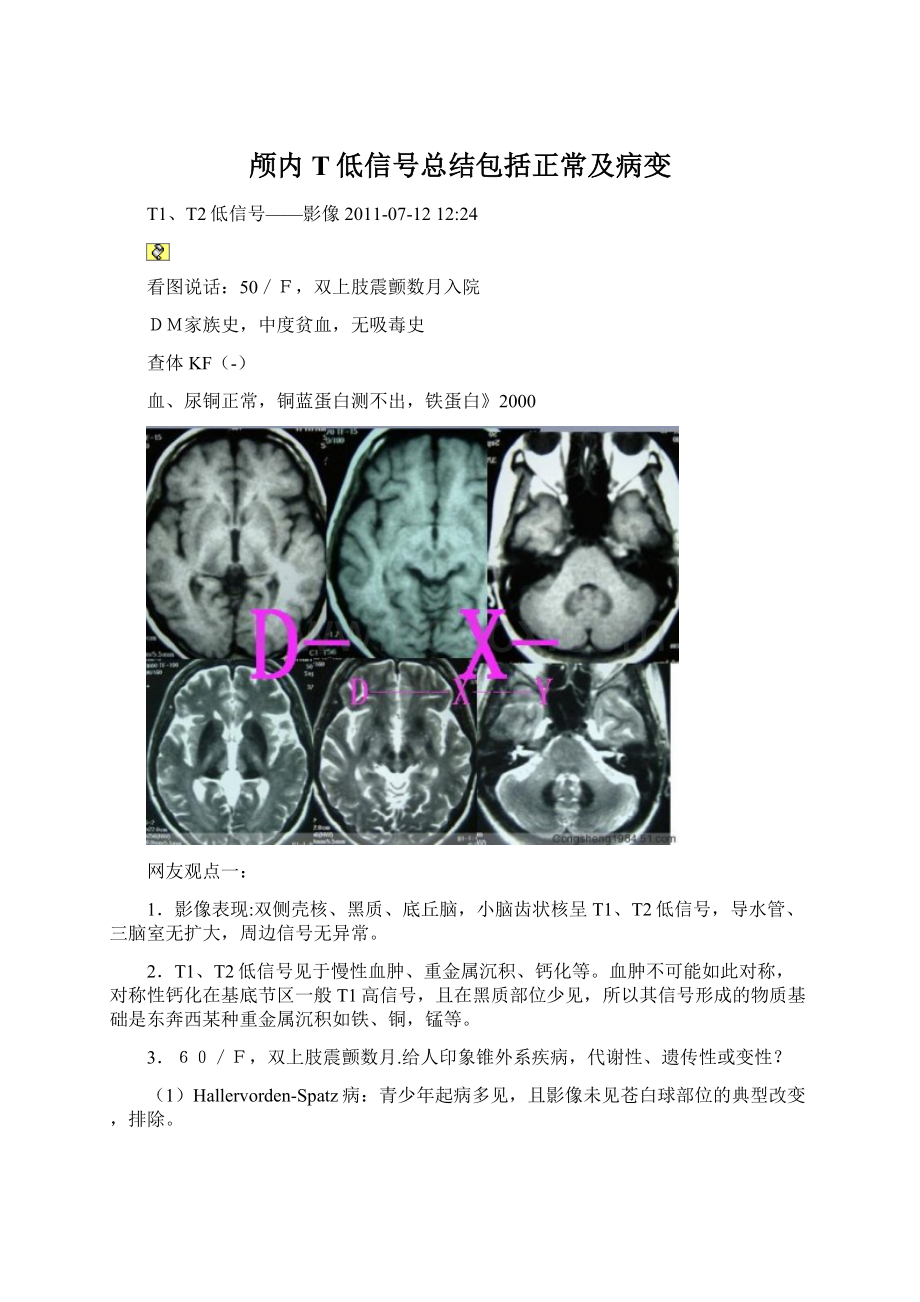
看图说话:
50/F,双上肢震颤数月入院
DM家族史,中度贫血,无吸毒史
查体KF(-)
血、尿铜正常,铜蓝蛋白测不出,铁蛋白》2000
网友观点一:
1.影像表现:
双侧壳核、黑质、底丘脑,小脑齿状核呈T1、T2低信号,导水管、三脑室无扩大,周边信号无异常。
2.T1、T2低信号见于慢性血肿、重金属沉积、钙化等。
血肿不可能如此对称,对称性钙化在基底节区一般T1高信号,且在黑质部位少见,所以其信号形成的物质基础是东奔西某种重金属沉积如铁、铜,锰等。
3.60/F,双上肢震颤数月.给人印象锥外系疾病,代谢性、遗传性或变性?
(1)Hallervorden-Spatz病:
青少年起病多见,且影像未见苍白球部位的典型改变,排除。
(2)慢性肝病性脑部变性:
其特征性影像基底节部位短T1,不支持。
但不完全排除,需明确有无肝病史,肝功能,血氨等。
(3)FAHR病:
因排除钙化可能,可排除。
(4)Wernicke脑病:
影像不支持,排除。
(5)肝豆:
年龄太大,排除
(5)原发性帕金森病:
(6)纹状体黑质变性:
因为随年龄增大,部分人可出现上述部位的铁质沉积。
在后二者中单凭此影像很难鉴别,看来得看临床症状及体征了。
文献:
Aceruloplasminemia
最初AEE称为家族性血浆铜蓝蛋白前体蛋白缺陷征[Miyajimaetal1987]或遗传性血浆铜蓝蛋白缺陷征[Yoshidaetal(1995)],AEE是一种常染色体隐性遗传性的疾病,表现为患者血浆内缺乏铜蓝蛋白和胰腺,肝脏及脑的铁质沉着.临床上表现为三联征:
视网膜变性,糖尿病和神经病变,发病年龄为25岁至60多岁.Miyajima等发现在***人非近亲结婚家庭中AEE的发病率大约为1/2,000,000.AEE是编码铜蓝蛋白的基因即CP基因发生突变引起的.CP基因位于染色体3q23-25区,大约有3.2kb,包括20个外显子.编码蛋白的铜蓝蛋白是一种蓝色的铜氧化酶,在脊椎动物中能携带大约95%血浆铜.在不同种族中45个患病家族中CP基因存在37种基因突变形式(21插入突变和16种错义突变),没有发现一种关键的突变形式,但在***人群中外显子15错义突变(2630位G变为A)相对多见,大约占到AEE等位基因的25~35%
异常基因产物
异常铜蓝蛋白被肝细胞释放后通常立即降解.CP基因错义突变后翻译的异常铜蓝蛋白被滞留在内质网内(早期分泌使途径),其它形式的突变的异常铜蓝蛋白减少进入高尔基体的铜含量,从而影响与铜蓝蛋白的结合。
铜蓝蛋白是一种关键的亚铁氧化酶,能催化二价铁转变为三价铁.这种疾病说明铜蓝蛋白在铁运输中起的重要作用.在AEE疾病中,铁的局部沉积与活动、储存与运输之间的平衡出现紊乱。
缺失铜蓝蛋白导致铁在各种脏器细胞内的异常沉积,包括胰腺、肝脏,视网膜和大脑基底节区。
铁沉积破坏这些脏器组织结构,引起AEE临床表现,通常发病于30岁到50岁之间。
临床表现
神经系统临床表现包括运动障碍(眼睑痉挛,扮鬼脸,颈项强直,震颤,舞蹈病)和共济失调(步态共济失调,发音因难)。
临床表现取决于铁在大脑沉积的部位。
AEE患者经常在发生糖尿病和神经系统病变前仅表现为贫血。
50岁以上患者中精神病学障碍包括抑郁和认知障碍
诊断及检查
在具有以上临床表现患者,诊断AEE依赖于血浆内铜蓝蛋白的缺乏和结合以下指标:
血浆铜含量降低;血浆铁含量降低;血清铁蛋白含量升高;肝细胞铁含量增高;在大脑(纹状体,丘脑,齿状核)和肝(T1,T2加权成像)铁沉积的MRI特征性异常低密度影的表现也支持诊断。
CP基因的遗传分子学检测仅运用于基础研究。
93%***患者中存在视网膜变性,视力不受影响。
许多散在黄斑及视网膜色素上皮细胞的淡灰色萎缩。
荧光素血管造影术表现的窗样缺损即为黄斑。
病理诊断:
内脏器官(特别是肝脏,胰腺和心脏)存在铁沉积。
肝脏不表现为肝硬化,铁在肝的含量比脑中多。
胰腺中胰岛b细胞具有铁沉积,导致糖尿病。
大脑铁含量分布依次为苍白球>壳>壳核>大脑皮层,小脑皮层。
铁在胶质细胞沉积比神经元中更突出。
鉴别诊断
大脑铁沉积性神经退行性变(Neurodegenerationwithbrainironaccumulation,NBIA)是一种进行性锥体外障碍性疾病,有脑部(主要基底节区)铁沉积的X线影像。
迟发性,缓慢性NBIA包括PKAN,这是一种由于PANK2基因突变引起的疾病。
Neuroferritinopathy,是一种由编码铁蛋白轻链基因发生突变引起的。
AEE,低血浆铜蓝蛋白不是它的特征。
铜蓝蛋白缺乏是一种铜代谢紊乱性疾病(肝豆状核变性,Menkes病)的特征。
肝豆状核变性是机体不能够使铜与铜蓝蛋白前体蛋白结合,从而胆汁分泌铜蓝蛋白减少,导致血浆中其含量缺乏而铜含量增加。
铜蓝蛋白缺乏是一种铜代谢紊乱性疾病(肝豆状核变性,Menkes病)的特征。
肝豆状核变性是机体不能够使铜与铜蓝蛋白前体蛋白结合,从而胆汁分泌铜蓝蛋白减少,导致血浆中其含量缺乏而铜含量增加。
以上特征,AEE显著不同于一种常见铁代谢疾病遗传性血色病。
因为AEE具有一些肝豆状核变性和HFE相关的遗传性血色病的特征,它可能会误诊为“轻度血色病伴低血浆铜蓝蛋白血症”或“轻度肝豆状核变性伴含铁血黄素沉着病”。
急性肝衰竭或任何原因导致失代偿肝硬化都能减少血浆铜蓝蛋白合成。
血浆铜蓝蛋白减少还可见于蛋白缺失性肠病,肾病综合征,营养不良。
治疗
去铁胺即一种铁鳌和剂,常用于血红蛋白浓度大于9克的有症状的患者,通过这种治疗可以降低血清铁蛋白浓度,减少铁在脑和肝脏的沉积,同时也能阻止神经系统症状的进展。
用法:
500mg去铁胺溶于100mL生理盐水静脉滴注一小时,每周两次,疗程6-10个月。
人新鲜冰冻血浆(FFP)静脉输注包含血浆铜蓝蛋白的FFP后,由于铜蓝蛋白的亚铁氧化酶活性使得血清铁的含量增加。
对于肝脏中铁含量的下降,联合应用FFP及去铁胺比单用FFP效果要好。
多次应用FFP后神经系统症状常能够得到改善。
其它抗氧化剂如维生素E、锌剂也可以和鳌合剂一起使用防治组织损伤,尤其是对肝脏和胰腺的损伤。
所有受累者从十五岁开始每年行糖耐量实验检查,糖耐量异常常是糖尿病开始的标志。
监测血红蛋白和糖化血红蛋白,当糖化血红蛋白>7.5%,血红蛋白>9g,/dL时开始应用去铁胺治疗。
最初的1-2个月500mg去铁胺每周一次静脉输注,患有AEE的患者很容易误诊为缺铁性贫血,并使用铁剂治疗从而加重铁的沉积
一、颅内具有短T2MR的生理结构
1.颅内正常生理结构
颅内有许多正常生理结构具有短T2MR征象,如颅骨内、外板、正常硬膜、正常血管的流空现象等,由于这些生理结构除呈短T2MR征象外,都具有长T1MR征象即T1加权或T2加权像都呈低信号,并且又都具有固定解剖部位,一般不会误诊,在此不作赘述,以下是几个特殊变异作些解释:
①岩骨穹隆所致引起小卵圆形短T2长T1MR信号,有时两侧对称出现,有时仅一侧出现,视患者扫描体位而定,勿误诊病变。
②鞍部前床突变异亦可引起上述同样MR改变,只有阅片时注意分析或加扫头部CT即可排除。
2.颅内含铁及钙的结构,特别是含有铁质的神经核团
(1)苍白球、壳核正常生理情况下即出现短T2MR征象。
(2)黑质及红核在一般情况下也出现短T2MR征象。
(3)小脑齿状核,在小脑半球中央呈横位卵圆形界线清晰的短T2结构。
中枢神经系统表面含铁血黄素沉积症在MR问世之前无法在活体身上检出,而MR问世以后,特别是在T2WI易显示信号降低。
铁在脑内代谢目前还不清楚,Drager研究证明:
出生后6个月在苍白球有少量铁沉着,黑质网状带则要12个月,红核则18~24个月,而齿状核则3~7岁见少量铁质沉着,在额部铁质沉着甚少,因而在T2WI上额部白质信号比枕部略低,随年龄增长,纹状体和齿状核铁质增加。
上述三种结构,一般在T1WI显示等信号,因为无论Fe2+或Fe3+都引起T2顺磁效应,缩短T2驰豫时间而T1改变则不明显,苍白球及壳核铁质沉着随年龄增长而增加,特别是60岁以上老年人,因而短T2征象亦随之明显。
这些结构又与所使用静磁场有关,磁场强度越高,短T2征象越明显。
3.正常髓鞘发育
出生二个月正常婴儿,由于脑白质特别是顶后部白质未髓鞘化而基底节区已髓鞘化,后者一般与灰质呈等信号,但由于与未髓鞘化高信号白质相对比,拟似短T2改变,要注意和熟悉小儿髓鞘化生理过程,否则容易误认为病变。
二、颅内具有短T2MR征象的病理改变
1.血管病变
动脉瘤,动静脉畸形,特别是无血栓形成情况下,均可产生短T2MR征象,这主要是血管流空决定的,即T1WI亦呈长T1低信号的流空效应,但由于T2WI有充足的脑脊液衬托显示更为清晰。
2.亚急性早期颅内血肿
即脑内出血3~7d内,这时正铁血红蛋白开始形成,但红细胞膜尚完整,由于具有磁敏感性,而T2WI呈低信号,而又由于正铁血红蛋白的形成,因此T1WI显示高信号。
3.钙化及骨化组织(脑膜瘤、听神经瘤、脉络丛乳头状瘤、少突胶质细胞瘤、骨瘤等)
凡是钙化或骨化组织都可产生短T2MR征象,即T2WI显示低信号。
钙化的形态、均匀与否,可产生均质或非均质短T2,但一般T1WI多显示等信号或呈长T1低信号,然而也有少数钙化病灶呈短T1高信号改变,注意识别。
4.富含脂肪组织(皮样囊肿、上皮样囊肿、畸胎瘤)
这类病变均含有不同的脂肪组织、类脂质、固态胆固醇或角蛋白,主要发生于中线、鞍上。
皮样囊肿一般呈短T2低信号,但也呈短T1高信号,一般是由于含脂肪的三磷酸甘油脂所致,且常常出现脂肪液平面。
而上皮样囊肿,很少呈短T2短T1的MR改变(因为很少含类脂质、三磷酸甘油脂),一般多呈长T1长T2改变,这是因为后者含有固态胆固醇及角化蛋白,不易产生驰豫的缘故。
畸胎瘤由于成份较复杂,亦含有不同量脂肪,能形成短T2MR征象,但信号不均匀可资鉴别。
脂肪成分与亚急性或慢性血肿及富含蛋白病变不易在T1WI作出鉴别,但T2WI一般都能作出鉴别。
因为:
脂肪组织在T2WI信号衰减,而蛋白含量高的病变则信号增高。
必要时作脂抑制成像可资进一步鉴别。
5.胶样囊肿
MR表现复杂,视囊内容物而定,可呈长T1或等T1信号,大约60%病例可呈短T1信号。
如果呈短T2短T1信号,即T1WI显示高信号T2WI显示低信号,则为陈旧性出血、液化胆固醇及囊内含有钙、铜、镁、钠、铁、磷等物质。
如果呈短T1长T2改变,即T1WI与T2WI均显示高信号,为粘蛋白及与出血有关。
如果呈长T1长T2改变则囊内富含浆液。
6.少见肿瘤如黑色素瘤
文献上认为黑色素瘤含有自由基而且有顺磁效应,因此呈短T2MR征象,但这种病变都含有短T1高信号为其特例不易误诊。
7.某些肉芽肿
一些炎性肉芽肿如脑结核、脑结节病,有时也出现短T2MR征象,但T1WI均为略低信号或等信号。
在一些手术病例中,组织学检查显示病变中央出现凝固性坏死,多系由凝固坏死所致。
8.异常铁及铜代谢疾病(病灶多对称)
(1)Pelizaeus-Merzbacherdisease(佩-梅病)
为性染色体遗传。
少突胶质细胞功能障碍,导致继发性髓鞘形成损害。
但临床及化验尚未发现特殊酶的不足,因此仅靠阳性家族史作出诊断。
临床表现包括头部震颤、眼球异常运动,通常在出生后3月前发病。
MR表现包括萎缩性改变,在T2WI显示正常灰白质信号倒置,提示正常髓鞘化障碍;同时,出现豆状核及丘脑短T2信号,可能是由于大量铁质沉着。
(2)Wilson病(肝豆状核变性)
主要为青年发病,为常染色体遗传疾病。
铜沉着于肝及脑引起肝硬化及神经症状,包括共济失调、震颤、痉挛。
脑铜沉着伴胶质细胞增生最显著在壳核,其次为皮层下、脑干、齿状核、黑质。
T2WI壳核信号减低,特别是用高场强MRI显像时。
这一顺磁效应是铜所致,但多数病例均呈长T2高信号,是铜与蛋白结合所致,与患者采用中低场MRI检查有关。
(3)Parkinson病
震颤麻痹是一种病因尚未完全清楚的锥体外系疾病,属慢性退行性病。
大多在中年之后发病,男性多见。
临床表现为肌张力增高、肢体震颤,主要病理改变在锥体外系的苍白球、黑质、壳核及尾状核,神经元减少萎缩、色素退化、胶质细胞增生及慢性少量出血。
高分辨MR图像显示脑干萎缩,累及黑质。
而T2WI信号减低,主要为铁质沉着于壳核、苍白球及黑质等。
(4)Shy-Drager综合征
亦为原因不明的退行性病变。
三角区的萎缩伴黑质及纹状体神经元消失、胶质细胞增生,类似Parkinson氏病改变;亦见于桥延髓核、小脑皮层、下橄榄。
MR表现为中脑、丘脑、基底节、三室周围短T2低信号伴黑质、苍白球铁质沉着。
(5)Hallervorden-Spatz病(哈-斯二氏病/苍白球色素性退变综合征)
大量铁质沉着于苍白球、黑质,较少见于大脑皮层;其它组织学变化包括神经元细胞肿胀、变性,特别是累及基底节以及髓鞘化异常。
铁代谢障碍的机制尚不清楚。
通常发生于学龄儿童。
有文献报导MR的T2WI苍白球信号降低,是由于铁质沉着,后者在高场强MRI显示更清晰,伴脑室周围信号增高。
9.r-刀治疗后
r-刀治疗的某些病是明显短T2长T1MR征象,即T1WI及T2WI都呈低信号,与高能量X线引起局部组织凝固坏死有关。


- 配套讲稿:
如PPT文件的首页显示word图标,表示该PPT已包含配套word讲稿。双击word图标可打开word文档。
- 特殊限制:
部分文档作品中含有的国旗、国徽等图片,仅作为作品整体效果示例展示,禁止商用。设计者仅对作品中独创性部分享有著作权。
- 关 键 词:
- 信号 总结 包括 正常 病变
 冰豆网所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
冰豆网所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
 对中国城市家庭的教育投资行为的理论和实证研究.docx
对中国城市家庭的教育投资行为的理论和实证研究.docx
-
二年级下册数学练习题大全.docx
-
二十年后回故乡的优秀作文.docx
-
软基换填施工方案.docx
-
《黑白装饰画》教案.docx
-
课堂教学改革实施方案5篇.docx
-
返璞归真简约致美解读《给予树》教学设计语文.docx
-
离职证明范本精选多篇.docx
-
《天局》全文.docx
-
我害怕作文集合15篇.docx
-
伏魔战记39详细攻略.docx
-
幼儿园学期计划.docx
-
雅思分类打印版Word格式文档下载.docx
-
年产1万吨竹子纤维加工项目可行性研究报告文档格式.docx
-
电商产业化项目投资经营商业计划书Word文件下载.docx
-
医学多媒体课件的设计与制作Word文档格式.docx
-
中学生中秋节想象作文Word格式.docx
-
等保20之漏洞扫描系统技术方案建议书Word文档格式.docx
-
培训学校个人工作计划模板5篇Word格式.docx
-
北京各区二模试题分类汇编文言文阅读Word文档下载推荐.docx
-
不同职业病危害因素的防护常识Word格式文档下载.docx
-
一年级上册同音形近字练习汇总Word文档格式.docx
-
班级家长会上班主任教师讲话稿Word下载.docx
-
科斯塔环载波恢复Word文件下载.docx
-
浙教义务版六年级语文下册教案 花潮Word文件下载.docx
-
集成电路设计与集成系统专业Word格式文档下载.docx
-
开工第一课专题讲座观后感文档格式.docx
-
东城区学年第一学期高三期末化学试题及答案Word格式文档下载.docx
-
苏教版六年级语文下册第七单元测试题Word格式文档下载.docx
-
学长征精神做红色传人活动方案文档格式.docx
-
读书笔记150字30篇文档格式.docx
-
中级经济法考前必背法条精华版备考资料Word格式.docx
-
学年高中生物第3章基因的本质第1节DNA是主要的遗传物质学案.docx
-
人教版四年级上学期数学教案二次备课及反思.docx
-
施工用电培训讲义doc.docx
-
学年人教版生物八年级上册54 细菌和真菌.docx
-
生鲜电商的市场分析报告.docx
-
学校安全教育主题活动设计.docx
-
人教版小学四年级音乐下册教案.docx
-
施工组织设计电梯前室.docx
-
阳离子交换器说明书.docx
-
实验学校1415学年下学期高二期中考试生物文试题附答案.docx
-
四川省泸州市泸州高级中学学年高一下学期第一次月考语文试题doc.docx
-
人教部编版语文三年级下册第二单元《7 鹿角和鹿腿》教学设计.docx
-
食品化学复习题与答案.docx
-
一级市政实务划的重点辅导.docx
-
市委全委扩大会上关于经济工作的讲话.docx
-
日本年轻人不想买房中国人抢日本房原因在这里.docx
-
市政工程助理工程师考试试题.docx
-
四年上册语文教案.docx
-
一年级语文下册第16课一分钟教学设计新课标人教版.docx
