 生物药物分析.docx
生物药物分析.docx
- 文档编号:30613565
- 上传时间:2023-08-18
- 格式:DOCX
- 页数:24
- 大小:386.11KB
生物药物分析.docx
《生物药物分析.docx》由会员分享,可在线阅读,更多相关《生物药物分析.docx(24页珍藏版)》请在冰豆网上搜索。
生物药物分析
生物药物分析
第八章抗生素类药物的分析
一、效价的测定方法:
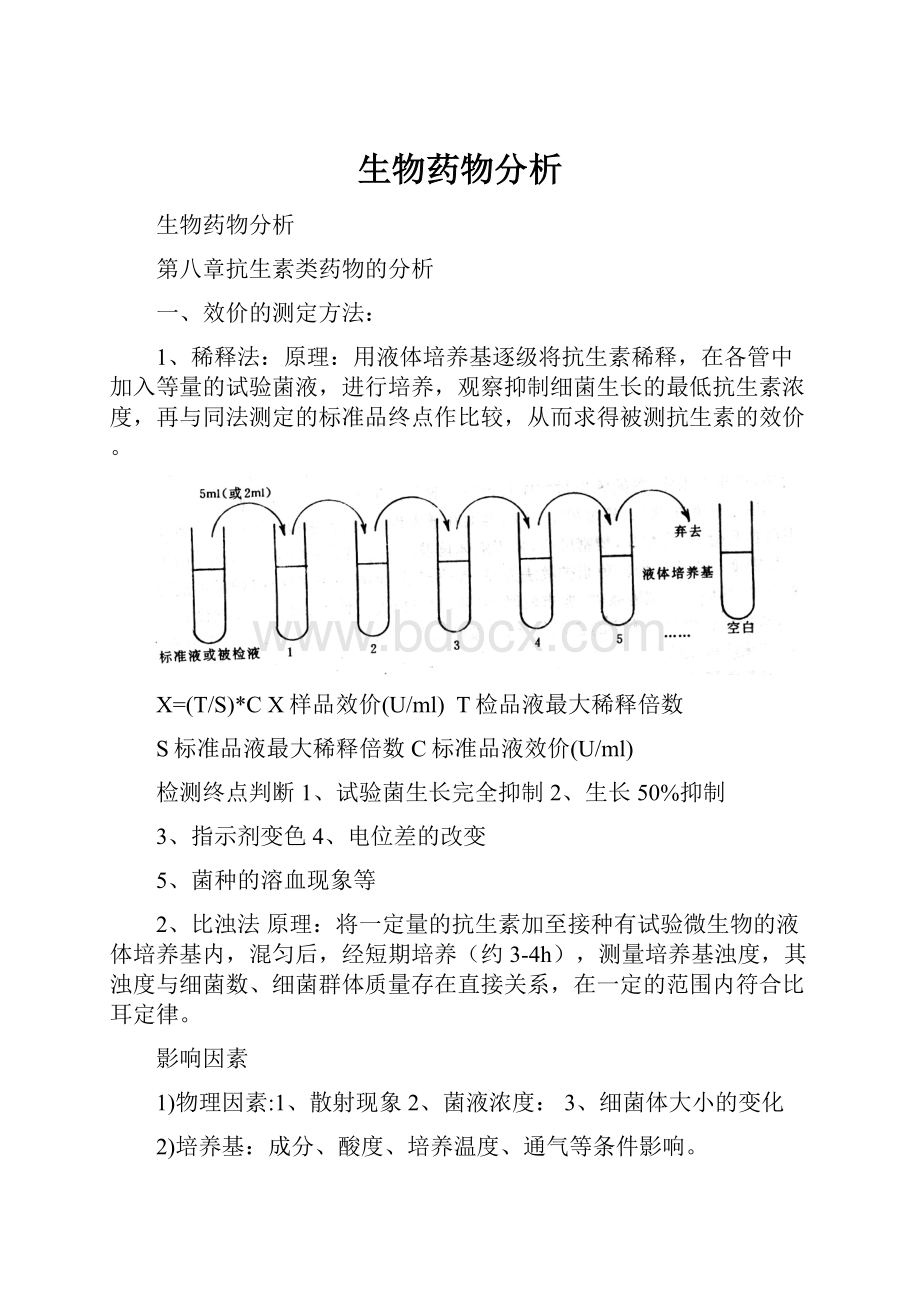
1、稀释法:
原理:
用液体培养基逐级将抗生素稀释,在各管中加入等量的试验菌液,进行培养,观察抑制细菌生长的最低抗生素浓度,再与同法测定的标准品终点作比较,从而求得被测抗生素的效价。
X=(T/S)*CX样品效价(U/ml)T检品液最大稀释倍数
S标准品液最大稀释倍数C标准品液效价(U/ml)
检测终点判断1、试验菌生长完全抑制2、生长50%抑制
3、指示剂变色4、电位差的改变
5、菌种的溶血现象等
2、比浊法原理:
将一定量的抗生素加至接种有试验微生物的液体培养基内,混匀后,经短期培养(约3-4h),测量培养基浊度,其浊度与细菌数、细菌群体质量存在直接关系,在一定的范围内符合比耳定律。
影响因素
1)物理因素:
1、散射现象2、菌液浓度:
3、细菌体大小的变化
2)培养基:
成分、酸度、培养温度、通气等条件影响。
金黄色葡萄球菌,pH在6.8-7.8。
3)培养:
温度、振摇。
3、琼脂扩散法即管碟法
原理:
抗生素在涂布特定菌的琼脂培养基内扩散,形成一定浓度抗生素的球型区,抑制试验菌生长,通过透明培养基观察抑菌圈,抗生素剂量的对数与抑菌圈面积或直径成正比。
在同样条件下将已知效价的标准品溶液与未知效价的供试品溶液的剂量反应(抑菌圈)进行比较;
当标准品和供试品是属于同一性质的抗生素时,标准品溶液和供试品溶液.对一定试验菌所得的剂量反应曲线,在一定剂量范围内应互相平行。
根据以上原理,可设计为一剂量法、二剂量法及三剂量法。
从而可以较为准确地测定供试品的效价
公式里各字母意义要记下。
影响抑菌圈半径的因素:
(1)D的影响因素:
抗生素本身结构、分子量等
杂质:
琼脂含量:
菌种对抗生素的吸附作用:
pH影响分子的形式、
温度影响扩散
D增大,r也增大
(2)T的影响:
一般T增加,r增大。
(3)M的影响:
在一定的范围内,小管内M越大,抑菌圈直径越大。
(4)C的影响:
C越小,抑菌圈直径越大
(5)H的影响:
培养基增厚,则抑菌圈减小。
操作步骤:
1、试验菌的纯化与悬液的制备
2、缓冲液、灭菌水与培养基的制备
3、标准品、供试品的称量、溶解与稀释
4、双碟的制备及放管
(二)薄层色谱
(三)光谱学鉴别
紫外光谱
红外光谱
六、利福霉素类抗生素(了解)
呈色反应:
1、浓盐酸反应:
2、三氯化铁:
向1ml利福霉素的溶液中,滴加三氯化铁乙醇溶液,有绿色,蓝绿色。
表明有酚羟基
3、硝酸盐反应:
利福平,溶液中加入硝酸钠溶液,颜色由橙色变为暗红色。
七、杂质检查
1、青霉素过敏原:
青霉噻唑多肽或青霉噻唑蛋白(青霉噻唑基团)
凝胶过滤法分离,用Penamaldate法测定
HgCl2
青霉噻唑衍生物Penamaldate衍生物(285nm)
2、链霉素
毒性杂质:
链霉胍,链胍双氢链糖;双氢链霉素;链霉素醛基化合物;
神经毒性:
二链霉胺
二链霉胺含量测定原理:
二链霉胺等不被KBH4还原,能与盐酸氨基脲反应,生成的缩氨脲在233nm有吸收峰
3、四环素
杂质:
脱水四环素、差向四环素、差向脱水四环素(HPLC测定)
4、土霉素
杂质:
脱水土霉素、差向土霉素、α-阿扑土霉素、β-阿扑土霉素等降解产物.(薄层分离,荧光测定)
第九章维生素及辅酶类药物分析
一、维生素C类的分析
1、性质
1)酸性:
烯二醇结构—酸性。
一元酸.能与NaHCO3作用生成钠盐
C3-OH,pKa=4.7(共轭效应)
C2-OH,pKa=11.57(邻位羰基)
2)还原性:
烯二醇基—还原性。
与硝酸银反应可生成金属银黑色沉淀
与2,6-二氯吲哚酚反应(2,6-二氯靛酚)
3)荧光反应:
4)糖的性质:
VC的结构很象糖结构,具有糖的性质。
可在三氯醋酸或盐酸存在下,经水解、脱羧、失水等反应,转变为糠醛,再与吡咯在50℃反应生成蓝色,用于鉴别反应
2、鉴别:
1)还原性:
维生素C分子中的二个烯醇基具有强还原性可以被氧化为二酮基
2)利用糖类的性质可在三氯醋酸或盐酸存在下,经水解、脱羧、失水等反应,转变成糠醛,再与吡咯在50℃反应生成蓝色。
3)紫外分光光度法:
根据维生素C在盐酸液(0.01mol/L)中,243nm有最大吸收
3、含量测定
1)碘量法:
维生素C由于分子中的烯二醇基具有还原性,能被I2氧化成二酮基.1mol维生素C与1molI2定量反应.终点颜色:
无色→蓝色。
反应条件:
酸性使VC在空气中氧化速度减慢。
溶剂新沸放冷的水。
减少水中溶解氧的影响
要排除亚硫酸的影响则在滴定前加入丙酮或甲醛。
2)2,6-二氯靛酚滴定法原理:
在酸性下,维生素C氧化型是红色的;还原型是无色的.酸性下直接用2,6-二氯靛酚滴定,用滴定剂自身的颜色变化指示终点,不需要另外的指示剂。
3)与1,10-菲洛啉-铁(III)试剂的比色法
基于维生素C将Fe(Ⅲ)还原生成Fe(Ⅱ),Fe(Ⅱ)与菲洛啉形成桔红色络合物,在510nm有最大吸收。
4)紫外分光光度法
二、维生素E的分析
合成型为dl-α-生育酚醋酸酯
天然型为d-α-生育酚醋酸酯
检查:
天然维生素E采用硫酸铈滴定法检查游离生育酚。
游离生育具有还原性,可被硫酸铈定量氧化
取本品0.10g,加无水乙醇5ml溶解后,加二苯胺试液1滴,用硫酸铈滴定(0.01mol/L)滴定(黄色),消耗硫酸铈滴定液(0.01mol/L)不得过1.0ml。
每1ml硫酸铈滴定液(0.01mol/L)相当于0.002154g的游离生育酚,维生素E中所含的游离生育酚的限量相当于不得过2.15%。
三、辅酶Q10的分析
1、鉴别
1)根据氧化型和还原型颜色不同来鉴别
氧化型辅酶Q10还原型辅酶Q10
(黄色)(无色)
2)氧化型辅酶Q10可将还原型亚甲蓝(无色)氧化而显蓝
3)紫外吸收光谱,275nm
第十章核苷酸类药物分析
一、嘌呤类核苷酸药物分析
1、鉴别
定糖法地衣酚orcinol反应:
核酸与浓盐酸共热时,可形成糠醛(绿色670nm)
二苯胺反应:
脱氧核糖核酸经酸水解后得脱氧核糖
(蓝色595nm)
间苯三酚反应:
其中的核糖在水溶液中加间苯三酚,水浴,显玫瑰红色
第十三章酶类药物的分析
一、酶活力测定方法
1)固定时间法
在适宜的条件下,使酶和底物共同保温一定时间,测定产物生成的量或底物消耗的量,计算出酶的含量(或活力)。
[E]表示酶浓度,[P]为反应产物浓度,t表示酶作用时间,K为常数。
2)连续监测法
在酶反应过程中,连续记录不同时间的底物消耗量或产物生成量。
(一)、需NAD+或NADP+作指示的连续监测法
还原型辅酶(NADH和NADPH)在340nm处有紫外吸收,氧化型辅酶(NAD+和NADP+)在340nm处无紫外吸收.利用340nm处每分钟吸收度升高或降低的速率与酶活性成正比的关系,推算出酶的活力单位。
包括:
直接测定法:
大多数需NADH(NADPH)参加的脱氢酶,可利用紫外分光光度法直接测定反应体系在340nm处吸收度的变化,计算酶活力单位。
丙酮酸+NADH+H+乳酸+NAD+
340nm处吸收度降低的速率与NADH的氧化速率成正比,与LDH的活力成正比。
偶联法:
此类酶催化反应不需NAD+或NADP+,但当与需NAD+或NADP+的脱氢酶反应偶联以后,能用紫外分光光度法测定.
由脱氢酶引起的反应为指示反应,脱氢酶是指示酶,指示酶活力要比测定酶活力至少大100倍。
例如:
谷草转氨酶的测定:
三联酶活测定:
引入一个辅助酶反应,将被测酶反应系统与指示酶反应系统联系起来,组成一个“三合一”酶反应系统,使底物转化率、辅助酶反应和NADH(NADPH)氧化速率之间成正比函数关系,辅助酶和指示酶的活力必须大大超过测定酶活力。
例如:
磷酸肌酸+ADP肌酸+ATP……
(1)
葡萄糖+ATPG-6-P+ADP……
(2)
G-6-P+NADP+
葡萄糖酸-6-磷酸+NADPH+H+…(3)
(1)被测反应,
(2)辅助反应,HK(己糖激酶)辅助酶,(3)指示反应,G-6-PDH(6—磷酸葡萄糖脱氢酶)指示酶。
(二)、不需NAD+或NADP+作指示的连续监测法
3)固定浓度法
根据酶催化反应,使反应产物达到额定的浓度时其反应时间与酶浓度成反比的原理进行设计的.[P]=K[E]t
以所需时间的倒数(1/t)对酶浓度作图即可制备标准曲线。
优点:
1、记录时间。
2、准确
二、酶活性测定方法的设计
一、正交设计——多因素选择
正交试验设计(Orthogonalexperimentaldesign)是一种高效的利用正交表来安排多因素多水平设计试验的方法。
通过巧妙的安排和分组,用较少的试验次数,就能分析各因素的作用大小,找出最佳的试验条件。
利用各因素所对应指标的极差R及平均极差D作出判断。
正交试验优选中性蛋白酶活力测定条件:
中性蛋白酶是一种蛋白水解酶,活力测定以酪蛋白为底物,水解产物与福林试剂在碱性情况下反应生成有色物质,于680nm处测定吸收值。
中性蛋白酶作用受缓冲液的pH值、底物浓度、反应时间及酶浓度等因素的影响。
供试品:
0.01%的中性蛋白酶溶液。
底物:
酪蛋白
实验方案
实验取四个因素:
PH值、底物浓度、反应时间、酶浓度。
每个因素选三水平,属于多因素多水平,为此选用L9(34)正交表进行实验。
正交试验表
正交实验安排表
各因素试验值计算结果
平均极差越大,对应的因素影响越大。
缓冲液PH值7.2,底物浓度0.5%,酶浓度用100U/ml为最佳反应条件。
反应时间为10,20,30min,对OD值无显著影响。
各因素试验值的方差分析
结论:
PH值(A)、底物浓度(B)、酶浓度(D)为非常显著因子;
反应时间(C)为非显著因子。
三、酶类药物的检测
1.肽键水解酶
1)胰蛋白酶:
由动物胰脏中提取的一种蛋白水解酶。
原理:
胰蛋白酶专一作用于赖氨酸、精氨酸等碱性氨基酸的羧基组成的肽键、酰胺键及酯键,水解速度为酯键>酰胺键>肽键。
BAEE(Benzoyl-L-Arginine-Ethyl-Ester)
BAA(Benzoyl-L-Arginine-Anide)
苯甲酰-L-精氨酸乙酯(BAEE)在胰蛋白酶的作用下,酯键被水解生成苯甲酰-L-精氨酸,在253nm波长处的吸收度随酶促反应递增,因此连续记录不同时间的产物生产量,根据活力单位定义计算酶活力。
测定法:
取呈直线的吸收度,按下式计算:
P为每mg供试品含胰蛋白酶的单位,U;A1为直线上终止的吸收度;
A2为直线上开始的吸收度;T为A1至A2读数的时间,min;
W为测定液中供试品的量,mg;
吸收度每分钟改变0.003,即相当于1个胰蛋白酶单位。
2)弹性蛋白酶
测定原理:
刚果红-弹性蛋白法:
以刚果红——弹性蛋白为底物,由于刚果红——弹性蛋白结合的共价键能被弹性酶水解,根据刚果红在495nm处有最大吸收,由标准曲线即可查得弹性酶单位数。
单位:
20分钟水解1mg刚果红-弹性蛋白所需的酶量为一个弹性酶活力单位。
方法:
3)尿激酶
效价测定原理(气泡上升法):
尿激酶激活人体内纤维蛋白溶酶原使其转化成纤维蛋白溶酶;纤维蛋白原在凝血酶的作用下,转变成纤维蛋白凝块,此凝块在纤维蛋白溶酶作用下,水解为可溶性小分子多肽。
在纤维蛋白溶酶原过量的情况下,尿激酶量与纤维蛋白凝块的溶解时间的对数成直线关系。
操作(气泡上升法)
试管中加纤维蛋白原溶液0.3ml(37℃水浴)
标准品(或供试品)1.0ml
加混合溶液0.4ml
立即摇匀计时,反应系统应在30~45秒内凝结,当凝块内小气泡上升到系统体积一半时作为反应终点。
以尿激酶的浓度为横坐标,以反应时间的对数为纵坐标。
b
2.脂键水解酶-胰脂肪酶(PancreaticLipase)
原理:
胰脂肪酶是一种水解酶,在一定条件下把甘油三脂类脂肪水解,最后生成甘油及脂肪酸。
底物:
橄榄油
用已知浓度的标准碱溶液滴定,可定量地测定脂肪酸的量,从而得知脂肪酶活力。
测定步骤
:
计算:
每分钟水解橄榄油产生1μmol脂肪酸的酶量,为1个活力单位。
每克含有的胰脂肪酶单位
A:
供试品消耗NaOH(0.1mol/L)的体积
B:
空白消耗NaOH(0.1mol/L)的体积
W:
供试品取样量(g)
N:
供试品稀释倍数。
3.糖苷键水解酶-溶菌酶
效价测定-比浊法:
以溶酶小球菌为底物,主要成分为粘多糖,粘多糖由NAG和NAM重复而成。
只能水解NAMC1和NAGC4之间的β-1,4糖苷键。
溶酶小球菌胞壁中的粘多糖经过溶菌酶的水解后,导致溶菌,溶液的吸收度下降。
在一定的条件下,每分钟吸收度下降0.001为一个酶活力单位。
计算:
酶活力单位(u/mg)=
W为测试液中供试品的重量(μg)
测定溶菌酶活性-比色法:
用染料艳红K-2BP标记的M.Lysodeikticus为底物,酶催化细胞壁分解时游离出染色碎片(产物)
反应后离心除去未分解底物,上清液比色,吸收度为溶菌酶活力的函数。
4.超氧化物歧化酶
效价测定:
SOD测定方法:
间接法:
使用氧自由基指示清除剂,SOD与指示清除剂竞争O2-,从而抑制指示清除剂与O2-的结合,根据指示清除剂与O2-反应速度的变化可间接测定SOD的活性。
包括:
黄嘌呤氧化酶-细胞色素C法和邻苯三酚法。
1)黄嘌呤氧化酶-细胞色素C法:
原理:
在有氧条件下,黄嘌呤氧化酶能催化黄嘌呤成尿酸,与此同时产生O2-,氧化型细胞色素C被O2-还原为还原型细胞色素C,后者在550nm有最大吸收,因此测定氧化性细胞色素C在加入SOD前后的光吸收变化可间接计算酶活性。
方法:
注意点:
在特定条件下,25℃(pH7.8)每分钟抑制细胞色素C还原速率达50%所需要的酶量为一个活力单位。
重金属离子易使黄嘌呤氧化酶失活,因此反应体系中必须添加EDTA。
反应体系中含过氧化氢酶可引起还原型细胞色素C发生过氧化作用,从而干扰检测的正确。
2)邻苯三酚法:
原理:
在碱性条件下,邻苯三酚会发生自氧化生成红
酚,同时生成O2-,当有SOD存在时由于它能催化O2-与H+结合生成O2和H2O2,从而阻止了中间产物的积累。
定义:
在一定条件下,每分钟抑制邻苯三酚自氧化速率达50%的酶量为一个酶活力单位。
操作:
定义:
在一定条件下,每分钟抑制邻苯三酚自氧化速率达50%的酶量为一个酶活力单位。
5.门冬酰胺酶
原理:
测定步骤:
在上述规定条件下,每分钟催化L-门冬酰胺水解释放1μg分子氨所需的酶量定义为1个酶活力单位.比活力测定方法:
采用Lowry法测定蛋白质含量,比活力=酶活力(U)/蛋白含量(mg)。
6.天冬氨酸酶活力的测定
一个酶活力单位定义为在测定条件下,每小时每克细胞转化生成1µmol天冬氨酸所需的酶量.
第十四章多糖类药物分析
一、多糖的分子量测定
1、凝胶色谱法:
将分子量不同的蛋白质通过一定孔径的凝胶固定相,由于各组分流经体积的差异,使不同分子量的组分得以分离。
最先淋出的是最大的分子。
Kd=0,Ve=V0;
Kd=1,Ve=V0+Vi;
0<Kd<1,Ve=V0+KdVi
2、粘度法
3、超离心法
4、高效液相色谱
色谱条件:
凝胶柱(分子量大小),示差折光检测器。
标准曲线:
标准曲线:
LogMW=a+btR
MW为重均分子量,tR为保留时间
待测多糖分子量(GPC专用软件)重均分子量Mw=∑(RIiMi)/∑RIi
Mi为供试品在保留时间ti时的分子量,RIi在第i部分中被洗脱物质的量(重量)。
二、多糖的含量测定
1、比色法(硫酸咔唑法、乙酰丙酮法、硫酸苯酚法、蒽酮法)
1)硫酸咔唑法:
己糖醛酸测定
在浓硫酸中,己糖醛酸与咔唑溶液反应生成的反应物呈红色。
520nm比色
2)乙酰丙酮法:
氨基己糖
氨基己糖在碱性条件下加热,可与乙酰丙酮缩合成吡咯衍生物,该衍生物与对-二甲氨基苯甲醛反应,反应物呈红色,于525nm测定吸收度。
3)硫酸苯酚法:
测定糖含量(总糖)
原理:
多糖在浓硫酸作用下水解成单糖,该单糖在强酸性条件下,与苯酚反应生成橙色衍生物。
在490nm处有最大吸收,其吸光度与浓度呈线性关系。
4)蒽酮法:
测定糖含量(总糖)
原理:
多糖在浓硫酸中水解后,进一步脱水生产糠醛类衍生物,与蒽酮作用形成蓝色化合物,620nm进行比色测定。
特点:
10-100μg范围内其颜色的深浅与糖含量成正比。
灵敏度高。
2、生物测定法(肝素钠)
3、生色底物法(低分子量肝素钠)
当蛋白酶(FⅩa)从生色底物分裂出pNA时,发色物的产生量与剩余的酶量成正比,与肝素效价成反比。
405nm测定。
三、多糖类药物结构分析
1、单糖组成
多糖组成单糖的分析
水解方法
(1)、完全酸水解法:
水解难易取决单糖性质及构型。
呋喃型戊聚糖较吡喃型己聚糖易水解,α型较β型易水解。
(2)、部分酸水解、碱水解:
选择温和的条件水解多糖,使糖链中某种类型的键特异性地打断
(3)、乙酰解:
多糖经过乙酰解反应(醋酐、冰醋酸)可生成乙酰化单糖和寡糖
(4)、甲醇解:
多糖链在80-100℃条件下与无水甲醇反应能将糖链变成组成单糖的甲基糖苷。
甲基糖苷能转化为三甲基硅醚衍生物或乙酰基衍生物,进行气相色谱分析
(5)、酶降解:
分为外切糖苷酶和内切糖苷酶
外切糖苷酶:
切下多糖非还原末端的一个单糖,并对单糖组成和糖苷键有专一性要求。
内切糖苷酶可水解糖链内部的糖苷键,释放多糖链片断以利于结构分析。
鉴定:
气相色谱法
高效液相色谱法
气相色谱法与质谱分析连用
2、糖苷键连接方式
1)红外光谱——糖苷键类型
确定吡喃糖糖苷键时,用红外光谱,在890cm-1处有特征吸收者,示有β-型糖苷键,在840cm-1处有特征吸收者,示有α-型糖苷键。
2)核磁共振谱——糖苷键(α型或β型)。
H-NMR谱中的化学位移δ5.4和δ5.1,有两个信号说明分子结构中的糖苷键为α型,如有δ4.53说明有β型。
3、糖苷键连接位置
(一)、高碘酸氧化、Smith降解
原理:
选择性的氧化降解反应,能够作用于多糖分子中1,2—二羟基和1,2,3—三羟基,生成过碘酸氧化产物;此产物用硼氢化钠还原后,再用酸水解,生成相应的醛、甲酸。
室温下用稀无机酸水解还原产物。
(二)、甲基化反应产物分析
(三)、乙酰解后质谱分析
第十二章多肽药物分析(该章节未划重点)


- 配套讲稿:
如PPT文件的首页显示word图标,表示该PPT已包含配套word讲稿。双击word图标可打开word文档。
- 特殊限制:
部分文档作品中含有的国旗、国徽等图片,仅作为作品整体效果示例展示,禁止商用。设计者仅对作品中独创性部分享有著作权。
- 关 键 词:
- 生物 药物 分析
 冰豆网所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
冰豆网所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
 《贝的故事》教案4.docx
《贝的故事》教案4.docx
-
《对韵歌》优秀教案8.docx
-
《函数yAsinωx+φ+P图象》wwwnet.docx
-
《静夜思》教学设计.docx
-
《汽车底盘构造与维修》题库与考核标准.docx
-
《世说新语》复习资料.docx
-
《我的服装我做主》教案设计.docx
-
《在品味情感中成长》教学片断设计.docx
-
11造价员《建设工程造价管理基础知识》精讲教程文件.docx
-
《不会叫的狗》教案 人教部编版1.docx
-
《操作系统》二学期A卷及答案.docx
-
《傅雷家书》名著阅读笔记.docx
-
《反不正当竞争法》下互联网平台封禁行为考辨以消费者用户合法权益保护为中心.docx
-
《化工原理》第六章蒸发.docx
-
《蓝海战略》概要11页.docx
-
《人生》读书心得.docx
-
《荷叶圆圆》公开课教案优秀教学设计26.docx
-
《科技出行研究报告》智能网联与新能源将变革未来汽车出行.docx
-
《272 向量的应用举例》导学案1.docx
-
《秋天》评课稿.docx
-
《电算化》第二章会计电算化的工作环境章节练习.docx
-
《室外给排水管道》施组.docx
-
《广东省建筑与装饰工程综合定额》计算规则.docx
-
《我多想去看看》教学.docx
-
《直通车车手基础认证》 考试答案 70题之欧阳育创编.docx
-
7天销量翻10倍皇冠卖家教您玩转最精准流量.docx
-
9 阿长和山海经.docx
-
《比例尺》教案.docx
-
《菜根谭》注译四闲适篇.docx
-
《福尔摩斯探案集》读后感15篇.docx
-
《红对勾》古代诗歌选择题答案补充.docx
-
《课堂密码》读后感及心得精选多篇.docx
-
高级高二下第二次联考化学试题定稿解析.docx
-
度测量仪器仪表量值溯源计划表.docx
-
高考考纲解读新版历史.docx
-
对加快西双版纳州新型工业发展的思.docx
-
反假币单选题试题及答案金储防伪2.docx
-
电工技术基础与技能课程标准.docx
-
鄂教版备考中考语文复习专题三形近字字音I卷.docx
-
防溺水安全责任书15篇.docx
-
高考英语模拟题分项汇编 专题03 完形填空第05期解析版.docx
-
二级建造师建设工程法规及相关知识重点记录重中之重.docx
-
电路图像分析题.docx
-
高频开关电源高频开关电源的维护.docx
-
房地产开发企业收入确认的条件及相关案例1.docx
-
高速公路新建项目招标分类和标段划分标准.docx
-
电容 电容品牌.docx
-
二年级下册数学易错题应用题带答案最新.docx
-
房屋倾斜度变形观测及内业处理报告.docx
-
发酵工程期末复习题教学内容.docx
-
电子秤电路测量与调试.docx
