 药物一致性评价培训班讲课要点.docx
药物一致性评价培训班讲课要点.docx
- 文档编号:30281552
- 上传时间:2023-08-13
- 格式:DOCX
- 页数:15
- 大小:233.91KB
药物一致性评价培训班讲课要点.docx
《药物一致性评价培训班讲课要点.docx》由会员分享,可在线阅读,更多相关《药物一致性评价培训班讲课要点.docx(15页珍藏版)》请在冰豆网上搜索。
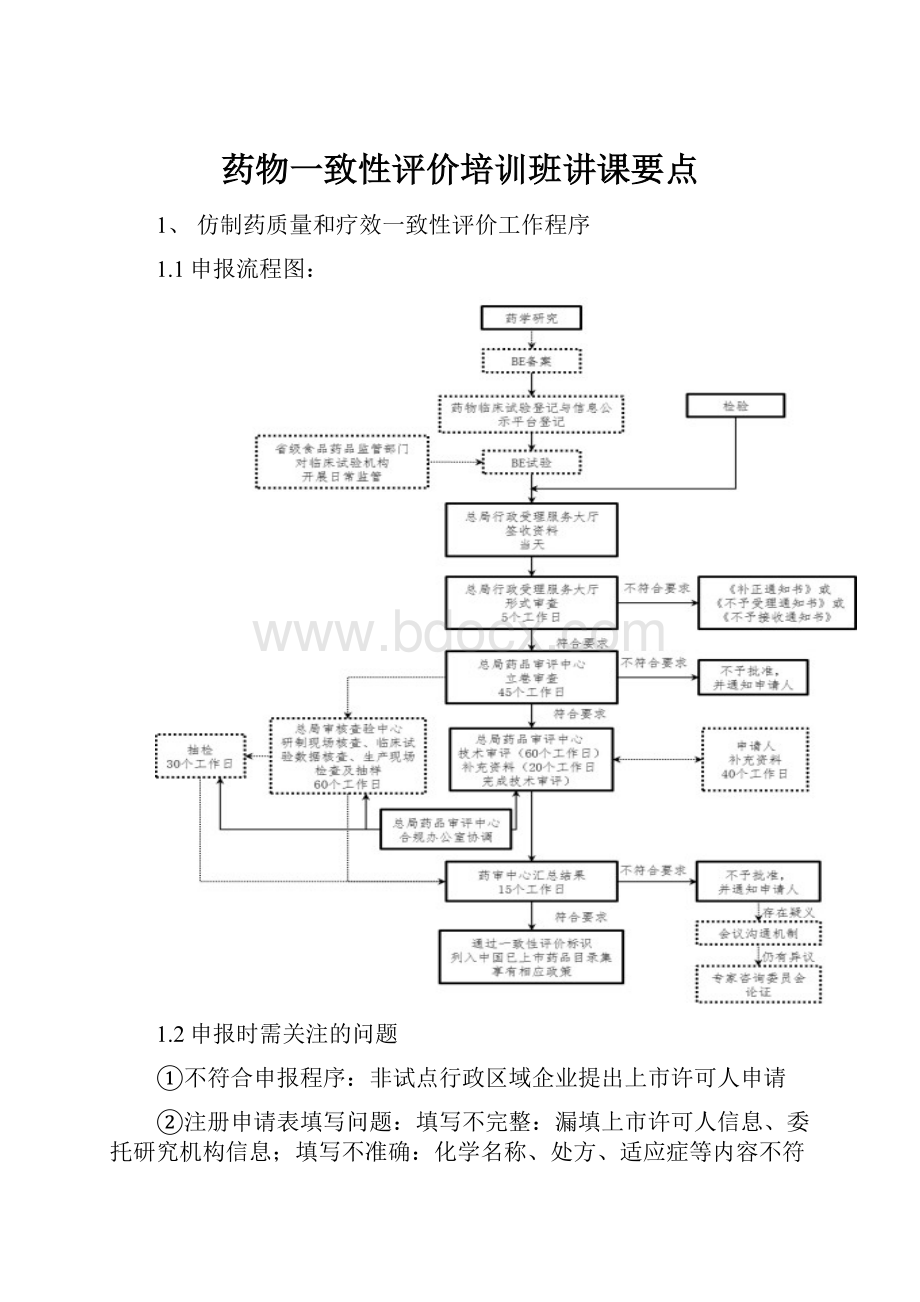
药物一致性评价培训班讲课要点
1、仿制药质量和疗效一致性评价工作程序
1.1申报流程图:
1.2申报时需关注的问题
①不符合申报程序:
非试点行政区域企业提出上市许可人申请
②注册申请表填写问题:
填写不完整:
漏填上市许可人信息、委托研究机构信息;填写不准确:
化学名称、处方、适应症等内容不符合要求;填写不一致:
申请表中规格、地址、标准编号、有效期等内容与证明性文件不一致
③申报资料问题:
资料项目不完整:
未按原注册批件要求提交相关资料,未提供相关证明性文件等
④邮寄方式错误:
补正资料收件人不应是个人,应按相关通知要求填写,同时应清晰注明资料内容
⑤注册联系人填写不准确:
未能准确填写申报资料联系人,导致电话联系效率低下
⑥电子文件提交不及时:
对于品种受理后应及时关注品种状态并按要求在申请人之窗提交电子文件
1.3相关的文件及指南
日期
工作文件
2016.5
总局关于落实《国务院办公厅关于开展仿制药质量和疗效一致性评价的意见》有关事项的公告(2016年第106号)
2016.5
总局关于发布仿制药质量和疗效一致性评价工作程序的公告(2016年第105号)
2016.6
总局关于推进仿制药质量和疗效一致性评价工作的通知(食药监药化管(2016)77号)
2016.8
总局关于发布化学药品仿制药口服固体制剂质量和疗效一致性评价申报资料要求(试行)的通告(2016年120号)
2017.8
总局关于仿制药质量和疗效一致性评价工作有关事项的公告(2017年第100号)
2017.9
总局关于发布《仿制药质量和疗效一致性评价受理审查指南(需一致性评价品种)》《仿制药质量和疗效一致性评价受理审查指南(境内共线生产并在欧美日上市品种)》的通告(2017年第148号)
日期
指导原则
2016.3
普通口服固体制剂参比制剂选择和确定指导原则
2016.3
普通口服固体制剂溶出曲线测定与比较指导原则
2016.3
以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则
2016.5
人体生物等效性试验豁免指导原则
……
2017.2
临床有效性试验一般考虑的通告
2017.2
改规格、改剂型、改盐基药品(口服固体制剂)评价一般考虑(3个技术指南)
2017.4
仿制药质量和疗效一致性评价品种分类指导意见
2017.5
仿制药质量和疗效一致性评价研制现场核查指导原则等
……
2、仿制药研发关键问题
①选择合理的参比制剂
临床价值品种:
国内外上市背景、安全性和有效性数据、上市后不良反应监测情况。
参比制剂:
是否具有完整和充分的安全性、有效性数据。
注射剂的选择(征求意见稿):
首选国内上市的原研药品;如原研药品国内未上市,应选择欧美日已上市的原研药品;在原研企业停止生产的情况下,可选择美国橙皮书标识为RS的药品。
②研发遵循质量源于设计(QbD)的理念
把被仿产品研究透,分析参比制剂的关键质量属性(CQAs):
如,理化性质、有关物质、溶出度(释放度)、含量、粒度、包封率等,设计试验方案。
③对原辅包的要求
共同审评:
制剂厂是责任主体。
共同审评可能引入新的问题:
变更来源、原辅包的相关变更。
原料药:
制剂生产商需结合原料药生产工艺,根据现有指导原则和相关文件对原料药的质量进行充分研究与评估,提高质量。
在后续的商业化生产中保证供应链的稳定。
辅料:
质量应符合要求。
包材:
相容性。
④制剂处方工艺:
处方:
依据是否充分,特别是早期上市产品;原辅料的相容性。
注射剂参考Q1、Q2的要求
工艺:
关键工艺参数的控制,工艺验证,无菌工艺验证。
包材:
注射剂的相容性研究。
批量:
考虑大生产的可行性,口服制剂、特殊制剂的放大效应。
常见的主要问题:
工艺明显不合理,如能耐终端灭菌的注射液采用F0值小于8的灭菌工艺;过量投料依据不充分;抑菌剂、抗氧剂的使用不当;批量小;关键工艺参数选择不当。
⑤质控方面:
标准的比较:
各国药典、已上市产品质量对比。
杂质研究:
杂质分析:
有关物质方法学研究的基础;结合原料药、处方工艺、相关文献进行分析,采用杂质对照品进行方法学验证。
原料药:
关注细胞毒杂质、元素杂质,参照ICH指导原则。
制剂:
重点关注降解产物。
与原研产品的对比研究:
杂质谱的对比、质量对比。
关键质量属性的研究:
原料药:
晶型、粒度等;制剂:
释放度、溶出度、粒径、包封率等。
常见的主要问题:
标准不完善:
杂质研究不全面:
没有对可能存在的杂质进行全面的分析和研究;对于超过鉴定限度的杂质没有进行研究;没有与参比制剂的杂质进行比较(杂质水平是否一致、是否有新杂质出现)。
关键质量属性研究缺失,如:
颗粒剂没有进行粒度的研究;口服混悬剂没有进行沉降体积比的研究。
⑥稳定性:
考察项目应全面,包括关键质量属性;对于半通透性的包材,关注湿度条件;贮藏条件的选择:
结合稳定性研究的结果选择贮藏条件,应与原研产品一致、药典一致。
常见主要问题:
考察指标设置不合理,未考察重要指标(如异构体、杂质、溶血磷脂等)。
放置过程中出现超过鉴定限度的未知杂质,未进行研究。
使用过程中的稳定性未考察,如稀释稳定性、配伍稳定性等。
⑦BE试验
BE评价要点:
伦理?
备案?
健康受试者or患者?
受试者样本量?
受试者基线情况是否一致?
空腹/餐后?
试验设计是否合理?
血药法、尿药法还是其他?
全血、血浆还是血清?
采样时长是否足够?
能否覆盖吸收、分布和消除三个时相?
采样点设计是否合理?
检测原形还是代谢产物?
提取方法是否合理?
分析方法是否合理?
方法学验证是否充分?
是否提供了完整的研究报告和支持性数据?
是否提供了全部色谱图?
色谱图信息是否完整?
PK参数采用何种软件,如何计算?
是否符合等效性评价标准?
是否存在其他问题?
常见的主要问题:
受试者入排标准执行不严格
受试者/患者管理(合并用药)
饮食管理不严格
周期间效应
参比制剂选择不合理
数据剔除或受试者脱落不合理
分批给药采血
采样点设置不合理
给药采血时间偏离
方法学验证不充分
生物样本分析质量控制不严格
生物样本分析方法不合理
复测原因或数据采纳不合理
制剂本身因素(原辅料、处方、工艺)
3、药物临床试验数据核查要点及规范性要求
3.1药物临床试验数据核查内容-样本检测
(1)仪器设备
关键实验设备、仪器应有相关运行、维护、维修记录,且时间与原始报告记录情况吻合。
检测用仪器是否开启稽查轨迹,轨迹有无时间逻辑性问题(修改电脑时间、进样时间间隔不当)。
(2)生物样本检测实验过程
完整的原始记录(包括实验单位、人员、日期、条件及实验结果等),时间、数据吻合。
生物样本分析方法学确证的原始数据与总结报告一致。
核查血药浓度数据与对应标准曲线计算的一致性;现场重新计算用以核实试验数据的真实性。
(3)生物样本管理
生物样本有接收、入库、存放、出库的原始记录,且记录完整(含样本标识、数量、来源、转运方式和条件、到达日期和到达时样本状态等信息)。
贮存的生物样本有领取、存入的原始记录。
在规定期限内,该项目保存的生物样本留样及其原始记录;核查留存生物样本的实际数量及记录的原始性。
(4)图谱溯源
图谱上的文件编码与受试者生物样本编码的对应关系能追溯源。
所有纸质图谱包含完整的信息(样品编号、进样时间、峰高/峰面积、血药浓度等)。
核查未知样本、方法学验证样本及随行标准曲线、QC样本的图谱,并在源计算机溯源,核对其与工作站电子图谱的一致性。
核查未知样本、随行标曲、QC样本图谱其进样/采集时间与文件编码顺序、试验时间顺序的对应一致性。
纸质图谱数据与总结报告一致性。
(5)其他
核查并记录影响Cmax、AUC等BE评价数据的手动积分图谱。
复测生物样本应有复测数量、复测原因、采用数据的说明。
血药浓度/药代动力学/生物等效性的分析计算数据及结果在相应的软件上可重现,且与总结报告一致。
3.2药物临床试验数据核查案例-检测部分
(1)分析测试系统无稽查轨迹:
生物样本分析测试仪器在本研究期间无稽查轨迹和工作日志,可随意修改或删除数据而不留记录。
(2)修改调换试验数据:
报告数据与原始记录数据不一致,如基质效应数据与源图谱数据计算结果不一致,原始记录中第四批标准曲线数据与图谱不一致;8名受试者多个时间点样品编号与对应的图谱中的文件名编码从小到大的顺序颠倒,原始记录和总结报告均未体现原因。
(3)瞒报修改试验数据:
检测原始记录本中的记录与稽查轨迹记录不符,方法学验证及生物样本测试的稽查轨迹中均多处出现分析测试系统日期反复更改、重复检测后用同一文件名命名并覆盖原有图谱,报告中仅提交其中一次文件的情况。
如:
2012年8月24日将系统日期调整为2012年8月16日,重新分析了全部4号和5号受试者的样本,其中4号受试者全部生物样本分析了3次,5号受试者全部生物样本分析了2次,最后报告只显示了最后一次的分析结果。
(4)原始记录缺失:
生物样本接收记录缺少样本标识、数量、转运方式和条件、样本状态等信息;贮存的生物样本无领取、存入的原始记录;生物样本检测原始实验记录中缺少每批次实验条件和过程、实验结果等信息的记录。
(5)分析测试过程不完整:
分析测试的关键实验设备、仪器没有相关维护记录;仪器使用记录不完整,缺少检测样品名称、数量等关键信息。
3.3临床样本分析的规范要求
(1)电子数据管理
原始电子数据与报告的一致性
归档数据(光盘)是否包括所有相关数据
失败分析批,样本重分析的数据
平衡仪器样品
稽查轨迹
数据备份,保存流程
数据修改等
(2)生物样本的管理
生物样本采集,储存,转移,运输
转移过程中的温控记录
生物样本有接收,入库,存放的原始记录,且记录完整,储存的生物样本有领取,存入原始记录
样本归档,销毁
dream做梦dreamed/dreamtdreamed/dreamt(3)实验记录的真实完整性
生物样本检测实验有原始记录且记录完整
sink下沉sank/sunksunk/sunken辅助记录是否与试验记录一致。
write书写wrotewritten生物样本分析方法学确证的原始数据与总结报告一致
血药浓度数据与对应标准曲线计算的一致性
(4)仪器设备管理
产生数据的设备和仪器应有定期维护维修记录
bear忍受boreborn与项目相关仪器设备的验证
日常使用需有使用日志;
计算机和工作站的稽查系统
(5)分析测试图谱的可溯源性
图谱上的文件编码/测试样本编码与受试者生物样本编码的对应关系能够溯源;
纸质图谱包含完整的信息(进样时间,峰面积,血药浓度等)
未知样本,标准曲线,QC样本的图谱,核对与原始电子图谱的一致性
lead引导ledled检查未知样本,随行标曲,QC样本图谱其进样/采集时间与实验时间顺序对应一致
纸质图谱数据与总结报告一致性
(6)样本重分析
样品重复分析原因,报告值选择
动词原形中文意思过去式过去分词样本重分析SOP和实际操作是否一致
cost花费costcost与重分析相关实验记录
ISR重分析,以及结果检查
(7)方法学验证
deal处理dealtdealt方法是否与样本分析是否一致
hear听到heardheard稳定性建立涵盖整个实验过程
prove证明provedproved/proven稳定性实验是否规范
残留测试
内标选择,基质效应
(8)未使用的电子数据
所有仪器上的数据应该有说明
不允许预打样本,标曲,质控
方法开发数据的保存,及实验记录
归档光盘包括项目产生的所有数据
数据不得删除或改动
4、省院复核检验
4.1、复核检验程序
(1)沟通咨询
需要由我院复核,一般需与相关业务科室沟通,预先审核药学研究内容,同时确定具体的复核方案;然后签订技术服务合同(包括复核项目、完成时间等)。
(2)收取样品
委托单位携带介绍信、申请委托函、修改完善的全套完整的技术资料;参比制剂1批和自制制剂3批(样品数量根据复核项目确定)送样复核检验。
按照药品检验机构质量管理体系的系列工作程序进行收检,在登记、编号并录入系统后下发至检验实验室,开启复核检验程序
4.2、复核检验技术指南各项目内容说明及实例
×××(品名)复核报告
一、摘要(600字以内)
根据所评价品种,进行综合阐述,国内外现状,取得批件的时间,生产质量情况等。
二、样品信息及申报资料审核
1.参比制剂信息
依照120号通告相关要求,全面、准确表述参比制剂基本信息,包括名称、生产企业、批次、批号、规格、获取渠道及购买相应凭证等。
明确参比制剂的选择是否与总局公布的参比制剂一致。
2.仿制制剂信息
详细、准确表述申请人所提供的仿制制剂基本信息,包括名称、生产企业、批次、批号、规格等。
明确仿制制剂在本次一致性评价工作中是否涉及处方工艺的变更。
3.申报资料审核
例:
XX制药有限公司于2008年取得5mg生产批件,批准文号为国药准字H20080599。
2010年取得10mg和20mg的生产批件,批准文号分别为国药准字H20103327、国药准字H20103548。
适应症为治疗抑郁症,治疗伴有或不伴有广场恐怖症的惊恐障碍。
此次申报单位一致性评价申报仿制制剂规格为5mg、10mg、20mg。
申报单位的三种规格产品均正常生产,5mg产品至2017年08月共生产287批,10mg规格至2017年08月共生产131批,20mg规格至2017年08月共生产30批,产品全部合格。
按国家食品药品监督管理局标准YBH10462008接受市场抽检三次,检验结果均合格。
本次申报处方工艺参照参比制剂处方工艺进行了重新开发,与原注册处方工艺比较:
……
申报单位提供了3个规格18批仿制制剂及3个规格的参比制剂的稳定性试验数据,目前加速试验已完成,长期试验正在进行中(已完成12个月)。
三、实验复核内容
1.关键质量属性考察
复核检测机构根据注册检验相关要求,按申报标准对申请人所提供的仿制制剂进行全项检验,对重点项目进行复核,并对申报标准提出复核意见。
复核内容:
(1)重点关注关键质量属性项目,如有关物质、含量、溶出度等;
(2)如果处方工艺发生了变更,质量标准重新修订,则需要进行方法学验证。
a.根据相关指导原则和企业提供的关键质量参数,同时对参比制剂和仿制制剂进行考察,重点关注申报单位的杂质谱研究。
b.溶出曲线的一致性复核(包括参比制剂与仿制制剂的一致性,仿制制剂批内、批间一致性等)
c.申报标准和样品检验结果的复核,标准的规范性,限度的合理性,检测结果是否与申报单位一致(提供三批检验报告)。
d.对于涉及处方工艺改变的品种,申报标准中是否规定了关键质量属性的质控项目,在实验复核的基础上对考察内容及方法进行评价或说明。
2.体外溶出研究
在进行溶出试验前,应按照《药物溶出度仪机械验证指导原则》的要求,对试验用溶出度仪进行机械及性能验证,并给出验证报告。
同时应关注企业申报资料中是否提供了试验用溶出度仪的机械验证报告。
按照《普通口服固体制剂溶出曲线测定与比较指导原则》(2016年第61号通告附件2)的要求,对参比制剂和仿制制剂的溶出曲线进行考察。
重点关注申报资料中溶出方法与检测方法的设计是否科学、合理,方法学考察数据是否完整、准确。
按照相关要求考察:
a.考察同一批次样品在不同溶出仪器上溶出曲线的差异;b.仿制制剂3批样品溶出行为的批内(n=12粒、片/批)和批间(n=3)均一性;c.复核检验结果与企业上报结果是否在分析方法允许的误差范围内;d关注企业是否按照《普通口服固体制剂溶出曲线测定与比较指导原则》要求,比较仿制制剂与参比制剂的溶出曲线相似性,复核检验的相似性结果与企业判断结果是否一致。
四、复核意见
例:
五、其他需要说明的问题
六、参考文献及相关实验数据


- 配套讲稿:
如PPT文件的首页显示word图标,表示该PPT已包含配套word讲稿。双击word图标可打开word文档。
- 特殊限制:
部分文档作品中含有的国旗、国徽等图片,仅作为作品整体效果示例展示,禁止商用。设计者仅对作品中独创性部分享有著作权。
- 关 键 词:
- 药物 一致性 评价 培训班 讲课 要点
 冰豆网所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
冰豆网所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
 《贝的故事》教案4.docx
《贝的故事》教案4.docx
-
《对韵歌》优秀教案8.docx
-
《函数yAsinωx+φ+P图象》wwwnet.docx
-
《静夜思》教学设计.docx
-
《汽车底盘构造与维修》题库与考核标准.docx
-
《世说新语》复习资料.docx
-
《我的服装我做主》教案设计.docx
-
《在品味情感中成长》教学片断设计.docx
-
11造价员《建设工程造价管理基础知识》精讲教程文件.docx
-
《不会叫的狗》教案 人教部编版1.docx
-
《操作系统》二学期A卷及答案.docx
-
《傅雷家书》名著阅读笔记.docx
-
《反不正当竞争法》下互联网平台封禁行为考辨以消费者用户合法权益保护为中心.docx
-
《化工原理》第六章蒸发.docx
-
《蓝海战略》概要11页.docx
-
《人生》读书心得.docx
-
《荷叶圆圆》公开课教案优秀教学设计26.docx
-
《科技出行研究报告》智能网联与新能源将变革未来汽车出行.docx
-
《272 向量的应用举例》导学案1.docx
-
《秋天》评课稿.docx
-
《电算化》第二章会计电算化的工作环境章节练习.docx
-
《室外给排水管道》施组.docx
-
《广东省建筑与装饰工程综合定额》计算规则.docx
-
《我多想去看看》教学.docx
-
《直通车车手基础认证》 考试答案 70题之欧阳育创编.docx
-
7天销量翻10倍皇冠卖家教您玩转最精准流量.docx
-
9 阿长和山海经.docx
-
《比例尺》教案.docx
-
《菜根谭》注译四闲适篇.docx
-
《福尔摩斯探案集》读后感15篇.docx
-
《红对勾》古代诗歌选择题答案补充.docx
-
《课堂密码》读后感及心得精选多篇.docx
-
涵桥施工方案.docx
-
工商管理论文题的目最新.docx
-
试验员考试试题及答案.docx
-
关于幼儿园家长会的相关工作汇报完整版.docx
-
汉字转化为拼音程序代码.docx
-
工业管道安装通用工艺规程.docx
-
售后服务部岗位职责.docx
-
行政工作计划范文.docx
-
观赏植物汇总表花期观赏特性栽植方式.docx
-
工艺管线焊接无损检测管理办法.docx
-
何家湾桥监理质量评估报告.docx
-
行政诉讼中的规范冲突与出路.docx
-
管理信息系统MBA教程之十培训体系搭建流程模版资料.docx
-
工作计划之乡镇应急预案编制计划.docx
-
杭州市学年中考一模物理试题A卷.docx
-
全考点C证安全员真题模拟考试.docx
-
河北三本排名.docx
-
人教版小学数学四年级下册单元提升练习题全册资料.docx
-
六年级年级组计划合集.docx
