 氨催化氧化制硝酸.docx
氨催化氧化制硝酸.docx
- 文档编号:28876639
- 上传时间:2023-07-20
- 格式:DOCX
- 页数:26
- 大小:264.33KB
氨催化氧化制硝酸.docx
《氨催化氧化制硝酸.docx》由会员分享,可在线阅读,更多相关《氨催化氧化制硝酸.docx(26页珍藏版)》请在冰豆网上搜索。
氨催化氧化制硝酸
三、氨催化氧化制硝酸硝酸和硫酸一样,也是无机化学工业中的重要产品,但它的产量比硫酸要小得多,1985年全世界的硝酸产量为3000万t/a,中国1993年的产量(以100%硝酸计)已达56.3万t/a。
硝酸大部分用来制造肥料,如硝酸铵、氮磷钾复合肥料等,亦大量用来制造炸药、染料和医药中间体、硝酸盐和王水等,还用作有机合成原料。
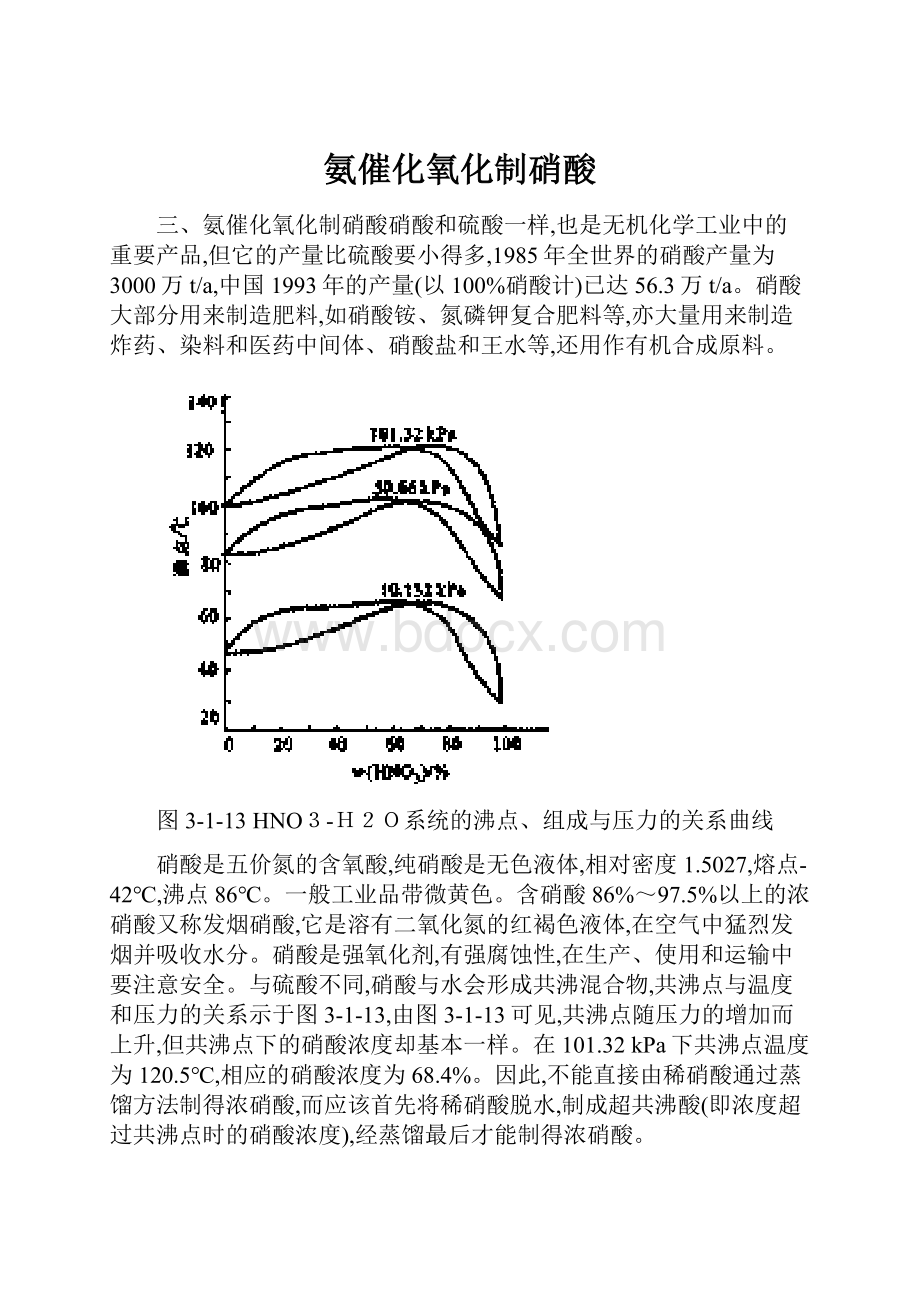
图3-1-13HNO3-H2O系统的沸点、组成与压力的关系曲线
硝酸是五价氮的含氧酸,纯硝酸是无色液体,相对密度1.5027,熔点-42℃,沸点86℃。
一般工业品带微黄色。
含硝酸86%~97.5%以上的浓硝酸又称发烟硝酸,它是溶有二氧化氮的红褐色液体,在空气中猛烈发烟并吸收水分。
硝酸是强氧化剂,有强腐蚀性,在生产、使用和运输中要注意安全。
与硫酸不同,硝酸与水会形成共沸混合物,共沸点与温度和压力的关系示于图3-1-13,由图3-1-13可见,共沸点随压力的增加而上升,但共沸点下的硝酸浓度却基本一样。
在101.32kPa下共沸点温度为120.5℃,相应的硝酸浓度为68.4%。
因此,不能直接由稀硝酸通过蒸馏方法制得浓硝酸,而应该首先将稀硝酸脱水,制成超共沸酸(即浓度超过共沸点时的硝酸浓度),经蒸馏最后才能制得浓硝酸。
1.生产方法综述
在十七世纪,人们用硫酸分解智利硝石(NaNO3)来制取硝酸。
硫酸消耗量大,智利硝石又要由智利产地运来,故本法目前已趋淘汰。
1932年建立了氨氧化法生产硝酸的工业装置,所用原料是氨和空气。
氨氧化催化剂是编织成网状的铂合金(常用铂-铑网),产品为稀硝酸(硝酸浓度为45%~62%)和浓硝酸(硝酸浓度为98%)。
(1)稀硝酸生产过程
A氨氧化主要反应有:
4NH3+5O2=4NO+6H2O
这是一个强放热反应。
反应温度760~840℃,压力0.1~1.0MPa,通过铂网的线速度大于0.3m/s,氧氨比(O2/NH3)为1.7~2.0,在以上工艺条件下,氨的氧化率可达95%~97%。
BNO的氧化出氨氧化反应器(亦称氧化炉)的反应气经废热锅炉和气体冷却器分出冷凝稀酸后,在低温下(小于200℃)利用反应气中残余的氧继续氧化生成NO2:
其中生成N2O3和N2O4的反应,速度极快(分别为0.1s和10-4s),而生成NO2的反应则慢得多(约20s左右),因此是整个氧化反应的控制步骤。
上列三个反应是可逆放热反应,反应后,摩尔数减少,因此降低反应温度,增加压力有利于NO氧化反应的进行。
NO的氧化程度α-NO与温度和压力
图3-1-14NO的氧化度α-NO与温度、压力的关系
的关系示于图3-1-14。
由图3-1-14可见,当温度低于200℃,压力为0.8MPa时α-NO接近100%,常压时α-NO也能达到90%以上,实际操作时α-NO在70%~80%之间,反应气即可送吸收塔进行吸收操作。
NO的氧化是一个非催化氧化反应,反应时间比氨氧化反应长得多,前者为20s左右,而后者仅为2×10-4s。
当氧氨比γ=1.7~2.0时,相应的氨浓度为11.5%~9.5%,为加速NO的氧化速度,此时需配入二次空气(它又可用作漂白塔的吹出气),将反应气中氧浓度控制在7.0%左右。
C吸收吸收在加压下进行。
氮氧化合物中除NO外,其他的氮氧化合物在吸收塔内与水发生如下反应:
因在常温下N2O3很容易分解成NO和NO2,因此由上列第三式生成HNO2的量不大,可以忽略不计。
上列各式生成的HNO2只有在温度低于0℃,以及浓度极小时方才稳定,在工业生产条件下,它会迅速分解:
3HNO2=HNO3+2NO+H2O-75.9kJ
因此,用水吸收氮氧化物的总反应式可写作:
3NO2+H2O=2HNO3+NO+136.2 kJ
即NO2中2/3生成硝酸酸,1/3变成NO,它仍需返回到氧化系统参与氧化反应,而且由于受共沸酸浓度的限制,硝酸浓度不会很高,一般在60%左右。
现有的稀硝酸生产方法有5种,即常压法、中压法(0.25~0.50MPa)、高压法(0.7~1.2MPa)、综合法(氧化为常压、吸收为加压)和双加压法(氧化为中压,吸收为高压)。
常压法氧化和吸收都在常压下进行,设备投资和动力消耗都较省,但制得的硝酸浓度不高,仅为45%~52%,排出的尾气中,氮化物NOx(NO、NO2及其他氮化物的总称)含量高,要增加处理装置,经治理后才能排入大气。
中压法、高压法和双加压法氧化和吸收都在加压下进行,设备投资和动力消耗大,但制得的酸浓度高,可达65~72%,尾气中氮化物NOx的含量比较低,容易处理或直接排放。
其中双加压法加压方式合理,吸收率达99.5%,尾气中NOx只有180ppm左右,可直接经烟囱排入大气,因此是值得大力推广的生产方法。
综合法氧化在常压下进行,吸收在加压下进行,设备投资和动力消耗介与常压和加压之间,硝酸浓度仍可达到65%~72%。
上述5种流程的消耗定额示于表3-1-08。
表3-1-085种生产方法消耗定额(以生产1吨100%硝酸计)
项目
常压法
中压法
高压法
综合法
双加压法
氨氧化压力/MPa(绝)
NO2吸收压力/MPa(绝)
氨[w(NH3)=100%]/吨
铂/克
电/度
冷却水/m3
副产蒸汽/吨
产品酸浓度/%
氧化率/%
吸收率/%
常压
常压
125
150
45-52
97
98
1
12
143
67
95-97
92
55-67
-
-
26
155
50
97
98
10
141
55-67
97
`
(2)浓硝酸生产过程生产浓硝酸有直接法、间接法和超共沸酸精馏法三种,直接法即由氨直接合成浓硝酸,基建投资较大,只在国内外大型工厂中采用。
间接法是先生产稀硝酸再设法将稀硝酸中水分脱除,将浓度提高至共沸酸浓度以上,最后经蒸馏得到98%成品硝酸。
这种生产方法适宜于中小型浓硝酸生产装置,在国内外采用比较普遍。
超共沸酸精馏法是使氧化气中水分脱除较多,使NOx直接生成超共沸酸,再经蒸馏制得浓硝酸的方法,被认为是制造浓硝酸的一种好方法。
A直接法生产过程直接法由氨和空气经氧化直接合成浓硝酸,生产的关键是除去反应生成的水。
反应经历以下五个步骤:
①制一氧化氮氨和空气通过铂网催化剂,在高温下被氧化成一氧化氮,并急冷至40~50℃,使生成的水蒸气经冷凝而除去。
②制二氧化氮一氧化氮和空气中的氧反应,生成NO2后,残余的未被氧化的NO和浓度大于98%的浓硝酸再反应,被完全氧化成二氧化氮:
③分出二氧化氮在低温下用浓硝酸(>98%)吸收二氧化氮成为发烟硝酸,不能被吸收的惰性气体(N2等)排出系统另行处理。
④制纯NO2并冷凝聚合为液态四氧化二氮加热发烟硝酸,它热分解放出二氧化氮,然后把这纯的NO2冷凝成为液态四氧化二氮:
⑤高压釜反应制浓硝酸将液态四氧化氮与稀硝酸混合(要求稀硝酸中水分与液态N2O4成一定比例)送入高压釜,在5.0MPa压力下通入氧气,四氧化二氮与水(来自稀硝酸)和氧反应直接生成98%浓硝酸。
为了加快反应的进行,加入的液态N2O4应比理论量多些,这样制得的是含大量游离二氧化氮(即发烟硝酸)的白色浓硝酸,将它放到漂白塔内,通入空气,把游离的NO2吹出,制到98%成品浓硝酸。
二氧化氮经回收冷凝后再送到高压釜使用。
如果氨的氧化不用空气,而采用纯氧(需加水蒸气稀释以防爆炸),制得的一氧化氮浓度可高些,这对以后的制酸操作是有利的。
但需建造制氧装置和增加动力消耗。
B间接法间接法所用脱水剂有硫酸、硝酸镁、硝酸钙和硝酸锌等。
经过多年生产实践的筛选,现在几乎全部采用硝酸镁。
图3-1-15Mg(NO3)2-H2O系统的结晶曲线
硝酸镁是三斜晶系的无色晶体,变成水溶液后,随浓度的不同,可以形成多种结晶水合物,图3-1-15示出了Mg(NO3)2-H2O系统的结晶曲线。
图中D点是临界溶解温度,即当硝酸镁溶液浓度为57.8%时,其结晶温度为90℃,此时析出Mg(NO3)2·6H2O结晶。
F点为转熔点,即当硝酸镁溶液浓度为81.1%时,其结晶温度为130.9℃,此时Mg(NO3)2和Mg(NO3)2·2H2O结晶共同析出。
因此在选择硝酸镁操作温度时,应该避开这些最高点,以免溶液结晶。
当硝酸镁溶液浓度大于67.6%时,其结晶温度随溶液浓度增加而迅速上升,溶液浓度超过81%时,则结晶温度直线上升,在此浓度下操作极易造成管道堵塞。
因此,硝酸镁浓度太稀脱水效果固然不好,太高则也难以操作,在实际生产中一般控制在64%~80%之间,即浓硝酸镁浓度不超过80%(一般为72%),加热器出口(即吸水后稀硝酸浓度)不低于64%。
硝酸镁法浓缩原理如下:
浓度为72%~74%的硝酸镁溶液加入稀硝酸中,便立即吸收稀硝酸中的水分,使硝酸浓度提高到68.4%以上,而硝酸镁由于吸收水分,浓度下降至65%左右,此时在硝酸和硝酸镁混合溶液的气相中HNO3浓度在80%以上,再将后者精馏即可得到成品浓硝酸。
Mg(NO3)2-HNO3-H2O三元混合物沸腾时所产生的HNO3蒸气成分可由图3-1-16求得。
从而确定浓缩硝酸所需的最低硝酸镁的用量。
图3-1-16Mg(NO3)2-HNO3-H2O三元体系气相中硝酸的平衡浓度
硝酸镁的脱水在真空下进行。
不同真空度下,硝酸镁水溶液的沸点见表3-1-09。
表3-1-09不同真空度下硝酸镁水溶液的沸点
品种
质量分数
%
真空度/kPa
沸点/℃
纯硝酸镁
含杂质的硝酸镁
-
-
-
-
-
*硝酸镁中的杂质主要有Ca(NO3)2,Al(NO3)3,Fe(NO3)3等
C超共沸酸精馏法该法由西班牙Espimdesa公司开发成功,技术关键是要求氨氧化反应后气体中水分要尽量除尽(冷凝酸浓度低于2%HNO3),使脱水后系统总物料中生成硝酸的浓度超过稀硝酸共沸点的浓度。
脱水反应气在氧化塔中用共沸酸氧化,使NO转化成NO2,在超共沸酸吸收塔中吸收生成浓度80%~90%的HNO3,再进入超共沸酸精馏塔、热酸漂白塔制得98%成品酸。
本法具有可大型化、投资省、运行费用低的优点,是目前最经济的方法,排出的尾气中NOx含量在200ppm以下,故可直接排放,不会造成环境污染。
现将三种浓硝酸生产方法可变成本的比较示于表3-1-10。
表中
(1)为超共沸酸精馏法;
(2)为全压法(压力0.45MPa)制取稀硝酸,再用硝酸镁法制浓硝酸;(3)为双加压法(氧化0.45MPa,吸收1.15MPa)制稀硝酸,再用硝酸镁法制浓硝酸。
由表3-1-10可见,超共沸酸精馏法可变成本最低,比全压法低118.24元/t,比双加压法低87.83元/t,对一个全年生产5万吨浓硝酸的工厂而言,超共沸酸精馏法可节约生产成本42~600万元/a。
表3-1-10三种浓硝酸生产的可变成本比较
2.氨的接触氧化原理
(1)氨氧化的化学平衡氨的接触氧化随反应条件和使用催化剂的不同可生成不同的产物:
除了上列反应外,还可能产生以下副反应:
氨的分解:
2NH3=N2+3H2-91.8kJ/mol
一氧化氮的分解:
2NO=N2+O2-180.3kJ/mol
氨和一氧化氮相互相用:
4NH3+6NO=5N2+6H2O+1804kJ/mol
(3-1-08)~(3-1-09)三个反应在900℃时的平衡常数,根据伦斯特(Nermst)公式计算结果如下:
所得平衡常数Kp1、Kp2、Kp3数值巨大,说明这三个反应实际上是三个不可逆反应。
其中Kp3特别巨大,若没有催化剂,氨氧化结果将主要生成氮气和水蒸气。
(2)氨氧化催化剂和催化机理采用的催化剂都为铂合金,常用的有Pt-Rh,Pt-Rh-Pd二元和三元合金,有的铂合金中还加进Co、Ni和Mo等金属以降低催化剂成本和减少铂在1500℃高温下的挥发性消耗。
通常所使用的铂丝直径为0.040~0.10mm,铂网上不为铂丝占据的自由面积是整个面积的50%~60%。
新铂网表面光滑而且具有弹性,活性小,为此需经活化处理。
方法是用氢气火焰进行烘烤,使之变得疏松、粗糙,从而增大反应接触表面积。
空气中的灰尘(各种金属氧化物)和氨气中可能夹带的铁锈和油污等杂质,会遮盖在铂网表面,造成催化剂暂时中毒,H2S也会使铂网催化剂暂时中毒,但水蒸气对铂网无毒害,仅会降低铂网的温度。
为此,原料气在混合前必须经过净化处理。
即使如此,催化剂在使用3~6个月后仍需进行再生处理。
再生方法是将铂网从氧化炉取出,浸泡在10%~50%的盐酸溶液中,在60~70℃下恒温1~2h,然后分离再用蒸馏水洗至中性,最后铂网经干燥和在氢气火焰中灼烧活化后,装入氧化炉重新参与反应。
氨催化氧化的反应机理,与SO2和O2在V2O5催化剂上的反应相仿,包括以下几个步骤:
①由于铂吸附氧的能力很强,铂催化剂表面吸附的氧分子中的共价键被破坏,生成二个氧原子;
②铂催化剂表面从气体中吸附氨分子,随后氨分子中的氮和氢原子与氧原子结合;
③进行电子的重新排放,生成一氧化氮和水分子;
④铂对NO和水分子的吸附能力较小,它们在铂催化剂表面脱附,进入气相中。
研究表明,气相中氨分子向铂网表面的扩散是整个催化氧化过程的控制阶段,也就是说整个反应是由外扩散控制的。
(3)氨催化氧化的反应动力学根据上述反应机理,М.И.捷姆金等导出800~900℃间在Pt-Rh网上的宏观反应动力学:
式中:
C0—氨空气混合气中氨的浓度,%;
C1—通过铂网后氮氧化物气中氨的浓度,%;
S—铂网的比表面积(活性表面cm2/铂网截面积cm2);
m—铂网的层数;
d—铂丝的直径,cm;
V0—标准状态下气体流量,L/h·cm2铂网截面积。
在实际生产中,C0,S,m,d是已知的,则通过(3-1-11)式就可求得在不同V0下的C1值,从而求得反应转化率X:
X=(C0-C1)/C0
X包括氨在主、副反应中的转化,因此要比前述的氧化率α-NO为大。
可利用下列公式直接计算出氨分子向铂网表面扩散的时间。
τ=Z2/2D(3-1-12)
式中:
Z—氨分子扩散途径的平均长度;
D—氨在空气中的扩散系数。
设氨在700℃下氧化,所用铂丝为0.009cm,1cm长的铂丝数为32根,则Z等于0.010cm,D等于1cm2/s,由(3-1-12)式可计算得扩散时间为5×10-4s。
实际操作中,反应温度在800℃左右,扩散时间还会缩短。
这一数据表明,氨氧化生成NO的反应速度是极快的,一般在10-4s左右时间内即可完成。
(4)氨催化氧化工艺条件的选择氨接触氧化的工艺,首先要保证有高的氧化率,这样可降低氨的消耗和硝酸的生产成本,常压下氧化率可达97%~98.5%,加压下也可达96%~97%;其次是应考虑有较高的生产强度和比较低的铂消耗,最大限度地提高铂网工作时间,从而达到操作的稳定性,生产的连续性。
A温度氨氧化生成一氧化氮虽在145℃时已开始,但到300~400℃时生成量仍旧很少,主要还是生成单质氮(N2)和水蒸气。
要使一氧化氮产率达到97%~98%,反应温度必须不低于780℃。
但反应温度过高,由于一氧化氮分解,一氧化氮产率不但不升高,还会有下降的可能,而且当反应温度高于920℃时,铂的损失将大大增加(主要是铂在高温下挥发加剧)。
一般氨在常压下催化氧化温度控制在780~840℃,加压下为870~900℃。
B压力从反应本身看,操作压力对于一氧化氮的产率没有影响,加压氧化(如在0.8~1.0MPa下操作)比常压氧化的氧化率还要低1%~2%,但铂催化剂的生产强度却因此而大为提高。
例如常压下每公斤铂催化剂每昼夜只能氧化1.5吨氨,而在0.9MPa下可氧化10吨氨,同一设备生产能力可提高5~6倍。
但压力过高,加剧了气体对铂网的冲击,铂网的机械损失(摩擦、碰撞后变成粉末)增大,因此一般采用0.3~0.5MPa,国外也有高达1.0MPa的。
C接触时间混合气体通过铂催化剂层的时间称为接触时间。
为保证氨的氧化率达到最大值,接触时间不能太长(即气流线速度太慢),因为这要降低设备的生产能力,而且氨容易分解成单质氮,使氧化率降低。
接触时间也不能太短,太短氨来不及氧化就离开铂催化剂层,同样会使氧化率降低。
生产实践证实,常压下接触时间以10-4s左右为宜,加压以1.55×10-4s左右为宜。
图3-1-17氨氧化率与氨空气混合气中氧氨比的关系
D混合气组成提高混合气中氧的浓度,即增加催化剂表面原子氧的浓度,不仅可强化氨氧化反应,而且也有利于一氧化氮氧化成二氧化氮。
但氨氧化反应加快,反应热增多,若温度控制不好,就会烧坏催化剂,甚至会酿成爆炸事故。
图3-1-17示出了混合气配比与氨氧化率的关系。
由图可见,直线1表示完全按生成一氧化氮反应时的理论情况,曲线2表示生产实际情况。
由曲线2可知,γ=O2/NH3达到1.7后,氧化率已递增不大,故一般将γ值维持在1.7~2.2之间,此时氧的过剩量比理论值约高30%。
不过,当反应温度较高时,例如达到800℃,反应速度已足够快,γ值可稍微低一些,取1.5~1.6。
若采用非铂催化剂,由于它的活性较小,γ值应大于2,以保持足够的氨氧化速度,否则氧化率会急剧降低。
根据γ值就可求得混合气体中氨的浓度在9.5%~11.5%范围。
E爆炸及预防措施当混合气中氨达到一定浓度时,可能会引起爆炸。
NH3-O2-N2混合气体的爆炸极限示于表3-1-11。
在混合气中通入水蒸气,可使爆炸极限范围变窄,甚至消失。
例如在混合气中通入10%以上的水蒸气时,在45℃的温度下已没有爆炸危险。
因此在生产中一般都加入一定量的水蒸气,这样即使将氨浓度提高到13%~14%也是安全的。
为防止爆炸,必须严格控制操作条件,使气体均匀地通过铂网;合理设计接触氧化设备;添加水蒸气;消除引爆隐患(如设备应良好接地,不用铁器敲击管路和设备,不穿带铁钉的鞋,车间不准吸烟等)。
表3-1-11NH3-O2-N2混合气的爆炸极限(以NH3%计)
O2+N2混合气中O2%
20
30
40
50
60
80
100
最低
最高
22
31
17
46
18
57
19
64
19
64
18
77
82
3.四种硝酸生产工艺流程
下面介绍四种硝酸生产工艺流程。
(1)双加压法制稀硝酸流程本法典型的工艺流程示于图3-1-18,现简述如下。
A氨的氧化和热能回收氨和空气分别进入过滤器,以除去气体中夹带的固体粉尘和油雾等对氨氧化催化剂有害的杂质,净化后的气体经混合器混合(混合气中氨含量约9.5%(v))后进入氨氧化器,经与铂铑网接触,96%~97%(v)的氨被氧化为一氧化氮,气体的温度也上升至~860℃,此气体经氨氧化器下部的蒸气过热器和废热锅炉回收热量后出氨氧化反应器的温度约为400℃。
图3-1-18双加压法制稀硝酸流程
BNO的氧化及吸收一氧化氮气体离开废热锅炉并经省煤器回收热量后,被冷却至约156℃。
当温度下降时,气体中的NO被氧化成NO2,然后进入水冷却器(Ⅰ),进一步冷却至40℃。
在这里,氧化氮(NOx)气体与冷凝水反应生成浓度约34%的稀硝酸。
酸气混合物经分离器分离,稀硝酸送入吸收塔。
由水冷器(Ⅰ)来的氧化氮气体,与来自漂白塔的二次空气相混合后进入氧化氮压缩机,被压缩至1.0MPa(表)。
气体经换热器被冷却至126℃,又经水冷却器(Ⅱ)进一步冷却至40℃后,氧化氮气体和冷凝酸一并送入吸收塔底部的氧化器继续氧化,在塔中氧化氮气体被水吸收生成硝酸,吸收塔的塔板上设有冷却盘管用以移走吸收热和氧化热,当塔内液体逐板流下时和氧化氮气体充分接触,酸浓度不断提高,在塔底部收集的酸浓度为65%~67%。
C漂白自吸收塔来的65%~67%的硝酸里溶入很多NOx气体,被送至漂白塔顶部,用二次空气将NOx气体从硝酸中吹出,引出的成品酸浓度为60%,含HNO2<0.01%,温度为62℃,经冷却至约50℃后,送往成品酸贮槽。
由吸收塔顶出来的尾气,经尾气预热器,被加热至约360℃,热气体进入尾气透平,可回收约60%的总压缩功,最后经排气筒排入大气。
排入大气的尾气中NOx含量约为180ppm。
(2)直接法制浓硝酸流程直接法制浓硝酸流程示于图3-1-19。
现简述如下:
图3-1-19直接法合成浓硝酸
A氨的接触反应和一氧化氮的初步氧化氨在铂催化剂上被氧化成氧化氮(主要为NO)进入氧化塔,与剩余的空气中的氧气氧化,生成二氧化氮,氧化率达90%以上。
氧化时发生的热量,用由发烟硝酸吸收塔上段(洗涤段)来的浓度约为65%的硝酸带走,硝酸被稀释至55%,送往混合罐。
B一氧化氮的再氧化和二氧化氮的吸收发烟硝酸吸收塔共分三段。
下段为重氧化段,气体中的一氧化氮在此被浓度为98%的硝酸几乎全部氧化成二氧化氮,同时硝酸被稀释至75%,中段为发烟硝酸吸收段,用被冷却到-10℃的98%浓硝酸作吸收剂,浓硝酸吸收二氧化氮后成为含30%游离NO2的发烟硝酸。
发烟硝酸由中段底部送至漂白塔,反应热用筛板上盘管中的冷冻盐水带走。
上段为洗涤段,以冷凝水洗涤尾气中的硝酸雾沫后成为65%稀硝酸,此酸送氧化塔。
尾气中还含有0.2%(2000ppm)的氧化氮,送至稀硝酸生产系统回收能量,并经治理后由烟囱排入大气。
C二氧化氮的解吸含30%游离NO2的浓硝酸至漂白塔,受热解吸,释放出NO2。
浓硝酸从塔底排出,经冷却后可供发烟硝酸吸收段循环使用。
塔顶出来的是纯二氧化氮,经初步冷却器用水冷却除去酸雾后,进入四氧化二氮冷凝器,用冷冻盐水冷凝为液态四氧化二氮,送至混合罐。
D合成浓硝酸混合罐中用液态四氧化氮和各处来的稀硝酸,配成N2O4∶HNO3∶H2O=7∶2∶1的混合物,在充分搅和的情况下用泵送至高压釜,在5.0MPa的压力和70℃下,混合物与纯氧反应生成浓硝酸。
送入高压釜中的N2O4是过量的,所以排出的浓硝酸中含有25%N2O4(称为热酸),送至漂白塔中部解吸,最后才成为成品酸(98%浓硝酸)。
(3)间接法生产浓硝酸流程间接法生产浓硝酸流程包括稀硝酸制造和稀硝酸浓缩二部分,前述5种稀硝酸生产方法都可与之配套。
图3-1-20示出的是稀硝酸的浓缩流程,用的脱水剂是硝酸镁。
现简述如下。
图3-1-20硝酸镁法浓缩稀硝酸工艺流程
72%~76%的浓硝酸镁溶液和稀硝酸按4~6∶1的比例在混合器7混合后,自提馏塔顶部加入。
该塔为填料塔,塔温115~130℃,所需热量由设在塔下部的加热器13提供。
含80%~90%HNO3蒸汽从提馏塔顶逸出进入精馏塔11中,该塔也是填料塔,塔顶逸出的是温度为80~90℃的98%HNO3蒸汽,经硝酸冷凝器1进入酸分配器9,在这里气体经风机23排出至稀硝酸系统,液体一部分(2/3)作回流,一部分(1/3)进漂白塔,经漂白后得到98%成品酸;由提馏塔底部流出的稀硝酸镁溶液进入加热器13。
用1.3MPa间接蒸汽加热,在174~177℃下脱硝(脱除NO2)。
产生的蒸汽用作提馏塔的热源。
出加热器13的稀硝酸镁浓度为62%~67%,用液下泵打入膜式蒸发器16进行蒸发。
经浓缩,硝酸镁提浓到72%~76%流入硝酸镁贮槽22中循环使用。
由膜式蒸发器出来的蒸汽,进入大气冷凝器15用水直接冷却,冷却水流入循环水池21,尾气用水喷射泵抽出,循环水池多余的酸水送废水处理系统。
近年来,中国的许多硝酸生产厂家对图3-1-20所示工艺作了改进,取得了良好的效果:
①将大气冷凝器15由直接冷凝器冷却改为间接冷凝冷却,废水量由原先的140~1


- 配套讲稿:
如PPT文件的首页显示word图标,表示该PPT已包含配套word讲稿。双击word图标可打开word文档。
- 特殊限制:
部分文档作品中含有的国旗、国徽等图片,仅作为作品整体效果示例展示,禁止商用。设计者仅对作品中独创性部分享有著作权。
- 关 键 词:
- 催化 氧化 硝酸
 冰豆网所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
冰豆网所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
 #2机组现场施工用电布置措施.docx
#2机组现场施工用电布置措施.docx
-
《个人贵金属质押借款合同》兴业银行.docx
-
《科学发展观和小康社会的经济建设》复习导学案.docx
-
《我和祖父的园子》第一课时教案两篇word.docx
-
《质量》教学案例与设计.docx
-
2惠农小册子.docx
-
7A版个人与团队模拟考试题及答案.docx
-
10篇新部编四年级下册语文课内外阅读理解专项练习题及答案.docx
-
16初四物理热和能知识点总结精讲.docx
-
20XX社会语言经典语录流行风暴.docx
-
48篇教学案例分析报告题.docx
-
《电子工厂安全管理制度汇总》.docx
-
《机械制造课程设计》指导.docx
-
《钱学森》教案第二课时.docx
-
《边城》读后感5篇.docx
-
《固定式压力容器安全技术监察规程》.docx
-
《论雷峰塔的倒掉》.docx
-
《手术台就是阵地》教学设计三年级语文下册.docx
-
《夏洛的网》课外阅读教学设计.docx
-
《自己的花是让别人看的》教案.docx
-
3C检查表090429.docx
-
7客运专线CRTSⅡ型板式无砟轨道施工工法.docx
-
《笔算除法》课时教案设计.docx
-
11#楼高大模板支撑体系专项方案.docx
-
17科学分析经济形势.docx
-
《电流和电路》易错题精讲综合检测题与答案.docx
-
《会计信息系统》习题含答案.docx
-
《汽车电器设备与维修》发电机分教考分离试题及标准答案.docx
-
《四川省排污许可证管理暂行办法》.docx
-
《新编实用英语》教案第一册Unit.docx
-
0母版锅炉值班员计算题WORD版.docx
-
3年级下册英语单词记忆人教版.docx
-
0623高一英语测试新世纪.docx
-
注册会计师税法重点不得抵扣进项税和进项税额转出每日一练1212.docx
-
关于眼界的议论文.docx
-
云南省某水库除险加固工程项目设计详细方案说明书报告.docx
-
孕妇贫血吃什么食物 吃这些食物补血好.docx
-
归个人使用及其具体用途在挪用公款罪中的定位.docx
-
英文优美一句.docx
-
Ezjmta011学习资料大全金融基础知识试题.docx
-
数字信号处理等波纹数字FIR低通滤波器.docx
-
《哈利波特》读后感.docx
-
浙江版高考地理第一讲 地球的宇宙环境和圈层结构.docx
-
浙江省杭州市高考语文命题比赛试题12.docx
-
《中级会计实务》之第十二章知识点总结.docx
-
海淀高三第一学期期中语文.docx
-
细胞培养资料.docx
-
PM集团固定资产管理制度.docx
-
投标书格式范本.docx
-
海事处优秀现场检查监督员的事迹材料.docx
-
浙江省温州市学年高一下学期期末语文试题.docx
