 σ键和π键.docx
σ键和π键.docx
- 文档编号:28080430
- 上传时间:2023-07-08
- 格式:DOCX
- 页数:13
- 大小:232.17KB
σ键和π键.docx
《σ键和π键.docx》由会员分享,可在线阅读,更多相关《σ键和π键.docx(13页珍藏版)》请在冰豆网上搜索。
σ键和π键
分子是由原子组合而成的。
是保持物质基本化学性质的最小微粒,并且又是参与化学反应的基本单元,分子的性质除取决于分子的化学组成外,还取决于分子的结构。
分子的结构通常包括两方面内容:
一是分子中直接相邻的原子间的强相互作用即化学键(chemicalbond),化学键的成键能量约为几十到几百千焦每摩;二是分子中的原子在空间的排列,即空间构型(geometryconfiguration)。
此外,在相邻的分子间还存在一种较弱的相互作用,其作用能约比化学键小一、二个数量级。
物质的性质决定于分子的性质及分子间的作用力,而分子的性质又是由分子的内部结构决定的,因此研究分子中的化学键及分子间的作用力对于了解物质的性质和变化规律具有重要意义。
化学键按成键时电子运动状态的不同,可分为离子键、共价键(包括配位键)和金属键三种基本类型。
在这三种类型化学键中,以共价键相结合的化合物占已知化合物的90%以上,本章将在原子结构的基础上着重讨论形成化学键的有关理论和对分子构型的初步认识,同时对分子间的作用力作适当介绍。
第一节现代价键理论
现代价键理论的基础
现代价键理论(valence bondtheory,简称VB法,又称为电子配对法)
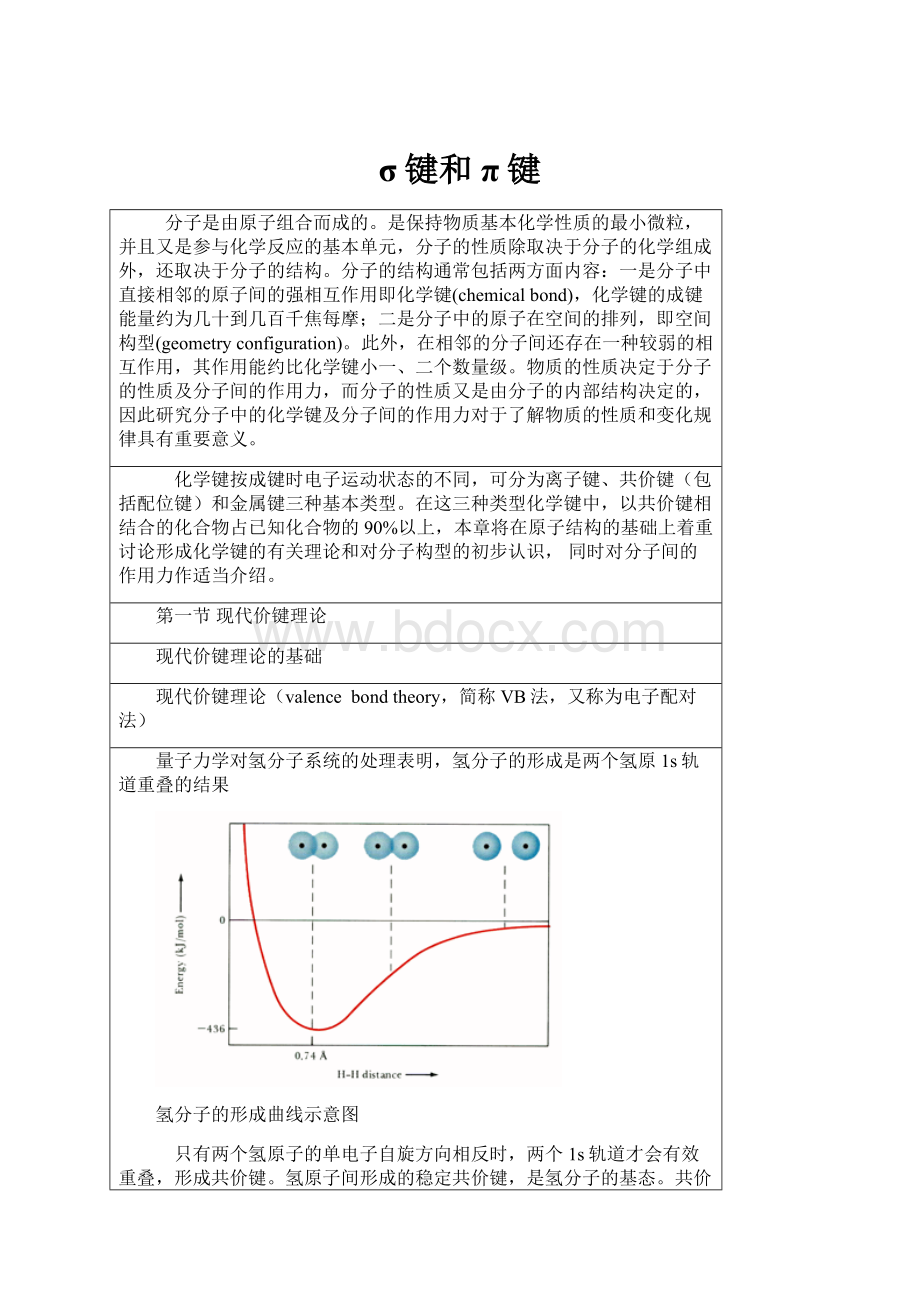
量子力学对氢分子系统的处理表明,氢分子的形成是两个氢原1s轨道重叠的结果
氢分子的形成曲线示意图
只有两个氢原子的单电子自旋方向相反时,两个1s轨道才会有效重叠,形成共价键。
氢原子间形成的稳定共价键,是氢分子的基态。
共价键的本质是电性的,但因这种结合力是两核间的电子云密集区对两核的吸引力,成键的这对电子是围绕两个原子核运动的,出现在两核间的概率较大,而且不是正、负离子间的库仑引力,所以它不同于一般的静电作用。
现代价键理论的要点:
1.两个原子接近时,只有自旋方向相反的单电子可以相互配对(两原子轨道重叠),使电子云密集于两核间,系统能量降低,形成稳定的共价键。
2.自旋方向相反的单电子配对形成共价键后,就不能再和其他原子中的单电子配对。
所以,每个原子所能形成共价键的数目取决于该原子中的单电子数目。
这就是共价键的饱和性。
3.成键时,两原子轨道重叠愈多,两核间电子云愈密集,形成的共价键愈牢固,这称为原子轨道最大重叠原理。
因此共价键具有方向性。
共价键特点:
共价键具有饱和性和方向性
如何理解共价键的饱和性和方向性?
例如:
在形成HCl分子时,H原子的1s轨道与Cl原子的3px轨道是沿着x轴方向靠近,以实现它们之间的最大程度重叠,形成稳定的共价键[图9-1]。
其他方向,因原子轨道没有重叠和很少重叠,故不能成键。
理论解释:
共价键的形成将尽可能沿着原子轨道最大程度重叠的方向进行。
原子轨道中,除s轨道呈球形对称外,p、d等轨道都有一定的空间取向,它们在成键时只有沿一定的方向靠近达到最大程度的重叠,才能形成稳定的共价键,这就是共价键的方向性。
根据要点1,只有自旋方向相反的单电子可以相互配对(两原子轨道重叠),形成稳定的共价键,因此共价键具有饱和性。
共价键的类型:
根据原子轨道最大重叠原理,成键时轨道之间可有两种不同的重叠方式,从而形成两种类型的共价键——σ键和π键。
σ键--以“头碰头”方式进行重叠,轨道的重叠部分沿键轴呈圆柱形对称分布,原子轨道间以重叠方式形成的共价键,
π键--原子轨道中两个互相平行的轨道如py或pz以“肩并肩”方式进行重叠,轨道的重叠部分垂直于键轴并呈镜面反对称分布(原子轨道在镜面两边波的瓣符号相反),而形成的共价键。
成键分析:
对于含有单的s电子或单的p电子的原子,它们可以通过s-s、s-px、px-px、py-py、pz-pz等轨道重叠形成共价键。
为了达到原子轨道最大程度重叠,其中s-s、s-px和px-px轨道沿着键轴即成键两原子核间的连线,如图9-4(a)所示,形成的共价键,这种共价键为σ键
σ键示意图,
(图9-1 σ键成键方式),当py-py、pz-pz以“肩并肩”方式进行重叠时,如图9-4(b)所示,也可形成共价键,这种共价键为π键
π键成键方式
(图9-2 π键成键方式),此时xy平面为对称镜面。
实例分析:
分析N2分子中化学键成分
N原子的电子组态为1s22s22px12py12pz1,其中3个单电子分别占据3个互相垂直的p轨道。
当两个N原子结合成N2分子时,各以1个px轨道沿键轴以“头碰头”方式重叠形成1个σ键后,余下的2个2py和2个2pz轨道只能以“肩并肩”方式进行重叠,形成2个π键。
所以,N2分子中有1个σ键和2个π键,其分子结构式可用N≡N表示。
由于σ键的轨道重叠程度比π键的轨道重叠程度大,因而σ键比π键牢固。
π键较易断开,化学活泼性强,一般它是与σ键共存于具有双键或叁键的分子中。
σ键是构成分子的骨架,可单独存在于两原子间,以共价键结合的两原子间只可能有1个σ键。
共价单键一般是σ键,双键中有1个σ键和1个π键,叁键中有1个σ键和2个π键。
因在主量子数相同的原子轨道中,p轨道沿键轴方向的重叠程度较s轨道的大,所以一般地说,p-p重叠形成的σ键(可记为σp-p)比s-s重叠形成的σ键(可记为σs-s)牢固。
正常共价键和配位共价键
根据成键原子提供电子形成共用电子对方式的不同,共价键可分为正常共价键和配位共价键。
正常共价键--如果共价键是由成键两原子各提供1个电子配对成键的,称为正常共价键,如H2、O2、HCl等分子中的共价键。
配位共价键--如果共价键的形成是由成键两原子中的一个原子单独提供电子对进入另一个原子的空轨道共用而成键,这种共价键称为配位共价键(coordinatecovalentbond),简称配位键(coordinationbond)。
通常为区别于正常共价键,配位键用“→”表示,箭头从提供电子对的原子指向接受电子对的原子。
实例分析:
分析CO分子中化学键成分
在CO分子中,O原子除了以2个单的2p电子与C原子的2个单的2p电子形成1个σ键和1个π键外,还单独提供一孤对电子( lonepairelectron)进入C原子的1个2p空轨道共用,形成1个配位键,这可表示为(CO图)
配位键必须同时具备两个条件:
一个成键原子的价电子层有孤对电子;另一个成键原子的价电子层有空轨道。
配位键的形成方式虽和正常共价键不同,但形成以后,两者是没有区别的。
关于配位键理论将在第十章配位化合物中作进一步介绍。
杂化轨道理论(hybridorbitaltheory)是1931年由PaulingL等人在价键理论的基础上提出,它实质上仍属于现代价键理论,但它在成键能力、分子的空间构型等方面丰富和发展了现代价键理论。
杂化轨道理论的要点:
1.在成键过程中,由于原子间的相互影响,同一原子中几个能量相近的不同类型的原子轨道(即波函数),可以进行线性组合,重新分配能量和确定空间方向,组成数目相等的新的原子轨道,这种轨道重新组合的过程称为杂化(hybridization),杂化后形成的新轨道称为杂化轨道(hybridorbital)。
2.杂化轨道的角度波函数在某个方向的值比杂化前的大得多,更有利于原子轨道间最大程度地重叠,因而杂化轨道比原来轨道的成键能力强。
3.杂化轨道之间力图在空间取最大夹角分布,使相互间的排斥能最小,故形成的键较稳定。
不同类型的杂化轨道之间的夹角不同,成键后所形成的分子就具有不同的空间构型。
轨道杂化类型及实例
按参加杂化的原子轨道种类,轨道的杂化有sp和spd两种主要类型。
按杂化后形成的几个杂化轨道的能量是否相同,轨道的杂化可分为等性杂化和不等性杂化。
sp型和spd型杂化
型杂化
能量相近的ns轨道和np轨道之间的杂化称为sp型杂化。
按参加杂化的s轨道、p轨道数目的不同,sp型杂化又可分为sp、sp2、sp3三种杂化。
(1)sp杂化
由1个s轨道和1个p轨道组合成2个sp杂化轨道的过程称为sp杂化,所形成的轨道称为sp杂化轨道。
每个sp杂化轨道均含有
的s轨道成分和
的p轨道成分。
为使相互间的排斥能最小,轨道间的夹角为1800。
当2个sp杂化轨道与其他原子轨道重叠成键后就形成直线型分子。
图9-3sp杂化过程及sp杂化轨道的形状
(2)sp2杂化
sp2杂化轨道的空间取向示意图
(图9-4 BF3的平面三角形构型和sp2杂化轨道的空间取向)
由1个s轨道与2个p轨道组合成3个sp2杂化轨道的过程称为sp2杂化。
每个sp2杂化轨道含有
的s轨道成分和
的p轨道成分,为使轨道间的排斥能最小,3个sp2杂化轨道呈正三角形分布,夹角为1200[图9-4]。
当3个sp2杂化轨道分别与其他3个相同原子的轨道重叠成键后,就形成正三角形构型的分子。
>>>>
一个分子的中心原子究竟采取哪种类型的轨道杂化,直接可以预测整个分子的空间构型。
杂化轨道理论成功地解释了部分共价分子杂化与空间构型关系,但是,仅用杂化轨道理论预测有时是难以确定的。
1940年美国的SidgwickNV等人相继提出了价层电子对互斥理论(valenceshellelectronpairrepulsiontheory),简称VSEPR法,该法适用于主族元素间形成的ABn型分子或离子。
该理论认为,一个共价分子或离子中,中心原子A周围所配置的原子B(配位原子)的几何构型,主要决定于中心原子的价电子层中各电子对间的相互排斥作用。
这些电子对在中心原子周围按尽可能互相远离的位置排布,以使彼此间的排斥能最小。
所谓价层电子对,指的是形成σ键的电子对和孤对电子。
孤对电子的存在,增加了电子对间的排斥力,影响了分子中的键角,会改变分子构型的基本类型。
根据此理论,只要知道分子或离子中的中心原子上的价层电子对数,就能比较容易而准确地判断ABn型共价分子或离子的空间构型。
价层电子对理论预测分子空间构型步骤为:
1.确定中心原子中价层电子对数
中心原子的价层电子数和配体所提供的共用电子数的总和除以2,即为中心原子的价层电子对数。
规定:
(1)作为配体,卤素原子和H原子提供1个电子,氧族元素的原子不提供电子;
(2)作为中心原子,卤素原子按提供7个电子计算,氧族元素的原子按提供6个电子计算;(3)对于复杂离子,在计算价层电子对数时,还应加上负离子的电荷数或减去正离子的电荷数;(4)计算电子对数时,若剩余1个电子,亦当作1对电子处理。
(5)双键、叁键等多重键作为1对电子看待。
2.判断分子的空间构型
根据中心原子的价层电子对数,从表9-4中找出相应的价层电子对构型后,再根据价层电子对中的孤对电子数,确定电子对的排布方式和分子的空间构型。
表9-4 电子对数和空间构型
实例分析:
试判断PCl5离子的空间构型。
解:
P离子的正电荷数为5,中心原子P有5个价电子,Cl原子各提供1个电子,所以P原子的价层电子对数为(5+5)/2=5,其排布方式为三角双锥。
因价层电子对中无孤对电子,所以PCl5为三角双锥构型。
实例分析:
试判断H2O分子的空间构型。
解:
O是H2O分子的中心原子,它有6个价电子,与O化合的2个H原子各提供1个电子,所以O原子价层电子对数为(6+2)/2=4,其排布方式为四面体,因价层电子对中有2对孤对电子,所以H2O分子的空间构型为V形。
表9-5理想的价层电子对构型和分子构型
实例分析6:
判断HCHO分子和HCN分子的空间构型
解分子中有1个C=O双键,看作1对成键电子,2个C-H单键为2对成键电子,C原子的价层电子对数为3,且无孤对电子,所以HCHO分子的空间构型为平面三角形。
HCN分子的结构式为H—C≡N∶,含有1个C≡N叁键,看作1对成键电子,1个CH单键为1对成键电子,故C原子的价层电子对数为2,且无孤对电子,所以HCN分子的空间构型为直线。
价键理论着眼于成键原子间最外层轨道中未成对的电子在形成化学键时的贡献,能成功地解释了共价分子的空间构型,因而得到了广泛的应用。
但如能考虑成键原子的内层电子在成键时贡献,显然更符合成键的实际情况。
1932年,美国化学家MullikenRS和德国化学家HundF提出了一种新的共价键理论——分子轨道理论(molecularorbitaltheory),即MO法。
该理论注意了分子的整体性,因此较好地说明了多原子分子的结构。
目前,该理论在现代共价键理论中占有很重要的地位。
分子轨道理论的要点:
1.原子在形成分子时,所有电子都有贡献,分子中的电子不再从属于某个原子,而是在整个分子空间范围内运动。
在分子中电子的空间运动状态可用相应的分子轨道波函数ψ(称为分子轨道)来描述。
分子轨道和原子轨道的主要区别在于:
(1)在原子中,电子的运动只受1个原子核的作用,原子轨道是单核系统;而在分子中,电子则在所有原子核势场作用下运动,分子轨道是多核系统。
(2)原子轨道的名称用s、p、d…符号表示,而分子轨道的名称则相应地用σ、π、δ…符号表示。
2.分子轨道可以由分子中原子轨道波函数的线性组合(linearcombinationofatomicorbitals,LCAO)而得到。
几个原子轨道可组合成几个分子轨道,其中有一半分子轨道分别由正负符号相同的两个原子轨道叠加而成,两核间电子的概率密度增大,其能量较原来的原子轨道能量低,有利于成键,称为成键分子轨道(bondingmolecularorbital),如σ、π轨道;另一半分子轨道分别由正负符号不同的两个原子轨道叠加而成,两核间电子的概率密度很小,其能量较原来的原子轨道能量高,不利于成键,称为反键分子轨道(antibondingmolecularorbital),如σ*、π*轨道。
3.为了有效地组合成分子轨道,要求成键的各原子轨道必须符合下述三条原则,也就是组成分子轨道三原则:
(1)对称性匹配原则
只有对称性匹配的原子轨道才能组合成分子轨道,这称为对称性匹配原则。
原子轨道有s、p、d等各种类型,从它们的角度分布函数的几何图形可以看出,它们对于某些点、线、面等有着不同的空间对称性。
对称性是否匹配,可根据两个原子轨道的角度分布图中波瓣的正、负号对于键轴(设为x轴)或对于含键轴的某一平面的对称性决定。
例如图9-10中的(a)、(b),进行线性组合的原子轨道分别对于x轴呈园柱形对称,均为对称性匹配;又如图9-11(d)和(e)中,参加组合的原子轨道分别对于xy平面呈反对称,它们也是对称性匹配的,均可组合成分子轨道;可是图9-11(f)、(g)中,参加组合的两个原子轨道对于xy平面一个呈对称而另一个呈反对称,则二者对称性不匹配,不能
组合成分子轨道。
图9-10原子轨道对称性匹配成键
符合对称性匹配原则的几种简单的原子轨道组合是,(对x轴)s-s、s-px、px-px组成σ分子轨道;(对xy平面)py-py、pz-pz组成π分子轨道。
对称性匹配的两原子轨道组合成分子轨道时,因波瓣符号的异同,有两种组合方式:
波瓣符号相同(即++重叠或--重叠)的两原子轨道组合成成键分子轨道;波瓣符号相反(即+-重叠)的两原子轨道组合成反键分子轨道。
图9-11是对称性匹配的两个原子轨道组合成分子轨道的示意图。
对称性匹配的两个原子轨道组合成分子轨道示意图
(2)能量近似原则
在对称性匹配的原子轨道中,只有能量相近的原子轨道才能组合成有效的分子轨道,而且能量愈相近愈好,这称为能量近似原则。
(3)轨道最大重叠原则
对称性匹配的两个原子轨道进行线性组合时,其重叠程度愈大,则组合成的分子轨道的能量愈低,所形成的化学键愈牢固,这称为轨道最大重叠原则。
在上述三条原则中,对称性匹配原则是首要的,它决定原子轨道有无组合成分子轨道的可能性。
能量近似原则和轨道最大重叠原则是在符合对称性匹配原则的前提下,决定分子轨道组合效率的问题。
4.电子在分子轨道中的排布也遵守Pauli不相容原理、能量最低原理和Hund规则。
具体排布时,应先知道分子轨道的能级顺序。
目前这个顺序主要借助于分子光谱实验来确定。
5.在分子轨道理论中,用键级(bondorder)表示键的牢固程度。
键级的定义是:
键级=(成键轨道上的电子数-反键轨道上的电子数)/2
键级也可以是分数。
一般说来,键级愈高,键愈稳定;键级为零,则表明原子不可能结合成分子。
分子间存在有vanderWaals力和氢键力。
这种分子间作用力的产生与分子的极化密切相关;分子的极化是指分子在外电场作用下发生的结构变化。
我们可以感受到这种作用力存在,如气体可以液化就是分子间存在相互作用力的最好证明。
分子的极性与分子的极化
分子的极性
根据分子中正、负电荷重心是否重合,可将分子分为极性分子和非极性分子。
正、负电荷重心相重合的分子是非极性分子;不重合的是极性分子。
对于双原子分子,分子的极性与键的极性是一致的。
即由非极性共价键构成的分子一定是非极性分子,如H2、Cl2、O2等分子;由极性共价键构成的分子一定是极性分子,如HCl、HF等分子。
对于多原子分子,分子的极性与键的极性不一定一致。
分子是否有极性,不仅取决于组成分子的元素的电负性,而且也与分子的空间构型有关。
例如CO2、CH4分子中,虽然都是极性键,但前者是直线构型,后者是正四面体构型,键的极性相互抵消,因此它们是非极性分子。
而在V形构型的H2O分子和三角锥形构型的NH3分子中,键的极性不能抵消,它们是极性分子。
分子极性的大小用电偶极矩(electricdipolemoment)量度。
分子的电偶极矩简称偶极矩(μ),它等于正、负电荷重心距离(d)和正电荷重心或负电荷重心上的电量(q)的乘积:
μ=q·d
其单位为10-30C·m 。
电偶极矩是一个矢量,化学上规定其方向是从正电荷重心指向负电荷重心。
一些分子的电偶极矩测定值见表9-5。
电偶极矩为零的分子是非极性分子,电偶极矩愈大示分子的极性愈强。
表9-5一些分子的电偶极矩
/10-30C·m
分子
分子
分子
H2
0
BF3
0
CO
Cl2
0
SO2
HCl
CO2
0
H2O
HBr
CH4
0
HCN
HI
分子的极化
无论分子有无极性,在外电场作用下,它们的正、负电荷重心都将发生变化。
如图9-13所示,非极性分子的正、负电荷重心本来是重合的(μ=0),但在外电场的作用下,发生相对位移,
图9-13 外电场对分子极性影响示意图
引起分子变形而产生偶极;极性分子的正、负电荷重心不重合,分子中始终存在一个正极和一个负极,故极性分子具有永久偶极(permanentdipole),但在外电场的作用下,分子的偶极按电场方向取向,同时使正、负电荷重心的距离增大,分子的极性因而增强。
这种因外电场的作用,使分子变形产生偶极或增大偶极矩的现象称为分子的极化。
由此而产生的偶极称为诱导偶极(induceddipole),其电偶极矩称为诱导电偶极矩,即图9-13中的△μ值。
分子的极化不仅在外电场的作用下产生,分子间相互作用时也可发生,这是分子间存在相互作用力的重要原因。


- 配套讲稿:
如PPT文件的首页显示word图标,表示该PPT已包含配套word讲稿。双击word图标可打开word文档。
- 特殊限制:
部分文档作品中含有的国旗、国徽等图片,仅作为作品整体效果示例展示,禁止商用。设计者仅对作品中独创性部分享有著作权。
- 关 键 词:
- 键和键.docx
 冰豆网所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
冰豆网所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
 《贝的故事》教案4.docx
《贝的故事》教案4.docx
-
《对韵歌》优秀教案8.docx
-
《函数yAsinωx+φ+P图象》wwwnet.docx
-
《静夜思》教学设计.docx
-
《汽车底盘构造与维修》题库与考核标准.docx
-
《世说新语》复习资料.docx
-
《我的服装我做主》教案设计.docx
-
《在品味情感中成长》教学片断设计.docx
-
11造价员《建设工程造价管理基础知识》精讲教程文件.docx
-
《不会叫的狗》教案 人教部编版1.docx
-
《操作系统》二学期A卷及答案.docx
-
《傅雷家书》名著阅读笔记.docx
-
《反不正当竞争法》下互联网平台封禁行为考辨以消费者用户合法权益保护为中心.docx
-
《化工原理》第六章蒸发.docx
-
《蓝海战略》概要11页.docx
-
《人生》读书心得.docx
-
《荷叶圆圆》公开课教案优秀教学设计26.docx
-
《科技出行研究报告》智能网联与新能源将变革未来汽车出行.docx
-
《272 向量的应用举例》导学案1.docx
-
《秋天》评课稿.docx
-
《电算化》第二章会计电算化的工作环境章节练习.docx
-
《室外给排水管道》施组.docx
-
《广东省建筑与装饰工程综合定额》计算规则.docx
-
《我多想去看看》教学.docx
-
《直通车车手基础认证》 考试答案 70题之欧阳育创编.docx
-
7天销量翻10倍皇冠卖家教您玩转最精准流量.docx
-
9 阿长和山海经.docx
-
《比例尺》教案.docx
-
《菜根谭》注译四闲适篇.docx
-
《福尔摩斯探案集》读后感15篇.docx
-
《红对勾》古代诗歌选择题答案补充.docx
-
《课堂密码》读后感及心得精选多篇.docx
-
动力拓展心得体会与动员讲话稿范文4篇汇编.docx
-
后勤服务专业安全操作规程.docx
-
20XX年采购部述职报告.docx
-
地理云南省蒙自市蒙自第一中学学年高一月考试题.docx
-
计算机操作员初级培训计划及大纲.docx
-
《异分母分数加减法》说课稿.docx
-
《平行四边形的认识》基础练习.doc
 网页设计与制作课程培训 -css基础.ppt
网页设计与制作课程培训 -css基础.ppt
-
《异分母分数加减法》前测分析第2组.doc
-
《年月日》教学设计与反思.doc
-
初二语文同步练习及答案.docx
-
贷款补充协议.doc
-
蚌埠市小升初数学模拟试题共10套详细答案.docx
-
高一物理上册月考检测试题085.docx
-
资中旅游网络营销策划案.doc
-
充分发挥小学民间体育活动的课题研究方案结题.docx
-
湖北省荆州市中考语文试题及答案Word版.docx
-
地下室施工专项方案.docx
-
八年级英语上册Unit5WildAnimalsPeriod5GrammarI教案新版牛津版.docx
