 第 4 章 GB2随机突变噬菌体文库的构建及淘选.docx
第 4 章 GB2随机突变噬菌体文库的构建及淘选.docx
- 文档编号:26082813
- 上传时间:2023-06-17
- 格式:DOCX
- 页数:20
- 大小:434.54KB
第 4 章 GB2随机突变噬菌体文库的构建及淘选.docx
《第 4 章 GB2随机突变噬菌体文库的构建及淘选.docx》由会员分享,可在线阅读,更多相关《第 4 章 GB2随机突变噬菌体文库的构建及淘选.docx(20页珍藏版)》请在冰豆网上搜索。
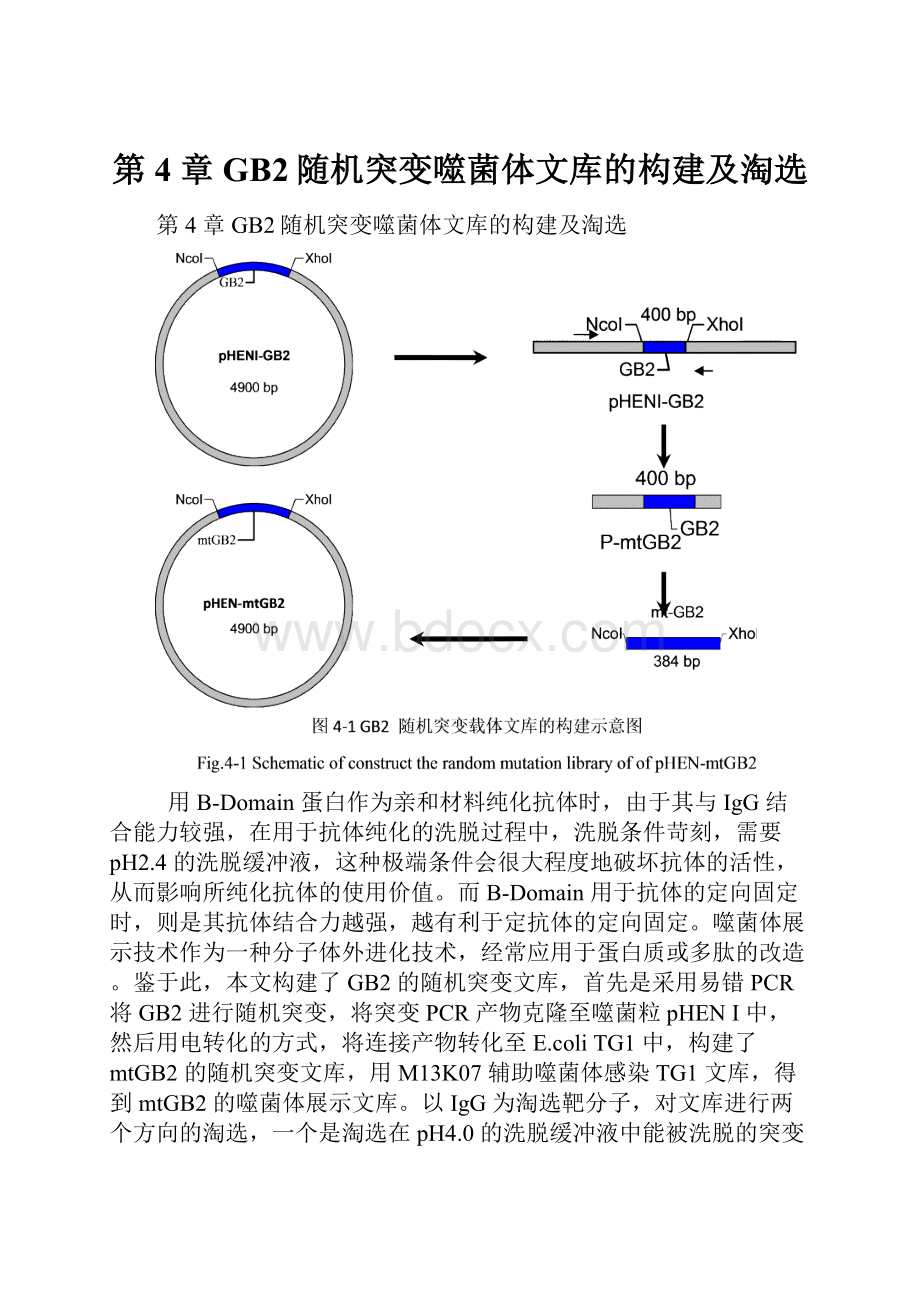
第4章GB2随机突变噬菌体文库的构建及淘选
第4章GB2随机突变噬菌体文库的构建及淘选
用B-Domain蛋白作为亲和材料纯化抗体时,由于其与IgG结合能力较强,在用于抗体纯化的洗脱过程中,洗脱条件苛刻,需要pH2.4的洗脱缓冲液,这种极端条件会很大程度地破坏抗体的活性,从而影响所纯化抗体的使用价值。
而B-Domain用于抗体的定向固定时,则是其抗体结合力越强,越有利于定抗体的定向固定。
噬菌体展示技术作为一种分子体外进化技术,经常应用于蛋白质或多肽的改造。
鉴于此,本文构建了GB2的随机突变文库,首先是采用易错PCR将GB2进行随机突变,将突变PCR产物克隆至噬菌粒pHENI中,然后用电转化的方式,将连接产物转化至E.coliTG1中,构建了mtGB2的随机突变文库,用M13K07辅助噬菌体感染TG1文库,得到mtGB2的噬菌体展示文库。
以IgG为淘选靶分子,对文库进行两个方向的淘选,一个是淘选在pH4.0的洗脱缓冲液中能被洗脱的突变型,能更好地应用于抗体纯化领域;一个是在pH2.4仍不能被洗脱的突变型,能更好地应用于抗体的定向固定。
图4-2亲和力减弱文库A淘选过程示意图
Fig.4-2SchematicofthepanningoflibraryA
图4-3亲和力增强文库B淘选过程示意图
Fig.4-3SchematicofthepanningoflibraryB
4.1材料、仪器与试剂
4.1.1材料
E.coliTG1购自Invitorgen、噬菌粒pHENI、HelpphageM13K07由英国剑桥医学研究委员会(MRC)分子生物学实验室GregWinter博士惠赠;小鼠腹水由本实验室制备。
4.1.2主要仪器
—BIO-RAD凝胶成像系统BIO-RAD公司
—低温离心机(2K-15)Sigma公司
—PCR扩增仪(GeneAmpPCRsystem2400型)PE公司
—型稳压电泳仪(DYY-IⅢ)北京六一仪器厂
—超低温冰箱(CD-196/HC)Thermo公司
—电转仪Bio-Rad公司
—电击杯Bio-Rad公司
—超纯水制备仪(67120)美国Millipore公司
—恒温摇床上海智诚
—96孔酶标板美国Costar公司
—37℃恒温孵育箱日本SANYO
—型全自动灭菌锅(MLS-3750)日本SANYO公司
—酶联免疫检测仪(MK2)Labsystems公司
4.1.3试剂
TaqDNA聚合酶、脱氧核糖核苷酸(dNTPs)、限制性内切酶(XhoⅠ、NcoⅠ、SalⅠ)、DL2000DNAmarker、λ-HindⅢDNAmarker,DNA片段纯化试剂盒、AgaroseGelDNAPurificationKitVer.2.0、质粒提取试剂盒均购买与TaKaRa公司;T4DNAligase购自NEB公司;Goldview核酸染料购自博大泰克生物基因技术有限责任公司;Yeastextract、Tryptone购自Oxoid公司;其他普通化学药品与试剂都是国产分析纯。
4.1.4溶液与培养基
1)LB:
在900mL蒸馏水中加10gTryton,5gYeastExtract,10gNaCl,溶解后加水至一升,用NaOH调节pH至7.4;
2)SOB培养基:
20g/LTrypone,5g/LYeastExtract,0.5g/LNaCl,高压冷却至
55℃后加入1mol/L的无菌MgCl210mL,250mmol/LKCl10mL;
3)SOC培养基:
除含有20mmol/L葡萄糖外,其他成分与SOB培养基相同;
4)SOB平板:
4g胰蛋白胨,1g酵母膏,0.1gNaCl,0.188gKCl,2.03g
MgCl2,0.986gMgSO4,3.6g琼脂粉,加ddH2O至200mL,高压灭菌,冷却后加入200µL100mg/mL氨苄青霉素,混匀后立即铺平皿,4℃保存备用。
5)2×YT:
在900mLddH2O中加16g胰蛋白胨,10g酵母膏,5gNaCl,加ddH2O至1L,调节pH至7.0,高压灭菌备用。
6)2×YT-G:
2×YT培养基中加入2%的葡萄糖;
7)2×YT-AG:
2×Y-GT培养基中加入100µg/mL的氨苄青霉素;
8)2×YT-AK:
2×YT培养基中加入100µg/mL的氨苄青霉素和50µg/mL的卡那霉素;
9)上层琼脂:
每升10g胰蛋白胨,5g酵母膏,5gNaCl,1gMgCl2.6H2O,7g琼脂粉,融化后分装试管3mL/管,高压灭菌,用时用微波炉融化,每加入10µL2M的MgCl2。
10)PEG-NaCl:
20%(w/v)PEG8000,称量200gPEG8000,146.1gNaCl,溶解于800mL去离子水中,定容至1L,高压灭菌,室温储存。
11)PBS:
称取NaCl8.0g,KH2PO40.2g,Na2HPO4·12H2O2.9g,KCl0.2g,加蒸馏水定容至1000mL。
12)洗涤缓冲液PBST:
称取NaCl8.0g,KH2PO40.2g,Na2HPO4·12H2O2.9g,KCl0.2g,加蒸馏水定容至1000mL,再加入Tween-200.5mL。
13)0.1MGly-HCl(pH2.4、4.0、6.0):
称取0.75gGly,溶解于与100mL去离子水中,用HCl溶液分别将pH调节至pH2.4、4.0、6.0。
14)1MTris-HCl(pH9.0)称取12.1gTris,溶解于去离子水中,定容至100mL,调节pH至9.0。
4.2实验方法与步骤
4.2.1野生型pHENI-wtGB2质粒的构建
1)合成引物
pHEN-GB2-NcoI:
GCACCATGGcaACTTACAAATTAATCCTTAATGG(NcoI)
pHEN-GB2-XhoI:
GACCTCGAGTTCAGTTACCGTAAAGGTCTTAGTC(XhoI)
pHEN-F:
CAGGAAACAGCTATGACC
pHEN-R:
GCCCCATTCAGATCCTCTTC
2)以pGEX-GB2为模板,pHEN-GB2-NcoI、pHEN-GB2-XhoI为引物进行PCR,扩增GB2片段。
3)分别NcoI/XhoI双酶切pHENI噬菌粒与PCR产物,分别割胶回收纯化。
4)用T4连接酶将GB2克隆至pHENI噬菌粒中,转化E.coliDH5a细胞。
5)菌落PCR、NcoI/XhoI双酶切验证所得克隆子,选取阳性克隆子送测序。
4.2.2GB2的随机突变
1)易错PCR对mtGB2进行随机突变,进行4组,每组的体系不同,分别降低一种dXTP的浓度至10%,引物为pHEN-F、pHEN-R,每管100μL体系,如下表:
表4-1易错PCR体系
Table4-1ThereactionsystemoferrorPCR
1
2
3
4
其他条件
﹡
﹡
﹡
﹡
dATP
0.02mmol/L
0.2mmol/L
0.2mmol/L
0.2mmol/L
dTTP
0.2mmol/L
0.02mmol/L
0.2mmol/L
0.2mmol/L
dCTP
0.2mmol/L
0.2mmol/L
0.02mmol/L
0.2mmol/L
dGTP
0.2mmol/L
0.2mmol/L
0.2mmol/L
0.02mmol/L
﹡pHENI-wtGB2:
0.2ng;MnCl2:
0.5mmol/L;MgCl2:
5mmol/L;Taq:
5U;上、下引物各20pmol。
反应条件:
94℃变性30s,62℃退火30s,72℃延伸30s,共35循环。
2)将4组易错PCR产物合并,用DNA片段回收试剂盒回收。
3)取部分回收产物进行TA克隆,挑取阳性克隆子送测序,分析随机突变效果。
4.2.3mtGB2与pHENI连接
1)用XhoI和NcoI两种限制性内切酶对mtGB2片段和pHENI噬菌粒进行双酶切,1.0%琼脂糖凝胶电泳后,用割胶试剂盒分别回收纯化,分别定量。
2)连接体系为50µL,按摩尔比5:
1投入GB2与pHENI,2.5µLT4连接Buffer,T4连接酶为0.5µL(350U),16℃反应过夜,共进行4管。
3)向连接产物中加入无水乙醇至终浓度为70%,-20℃沉淀过夜,4℃,10000rpm离心20min,去上清,用70%乙醇洗涤沉淀两次,待乙醇除干净后将沉淀溶解于20µL的无菌水中。
4.2.4电转感受态细胞的制备
1)从LB平板上挑取一个单菌落,接种于5mL的SOB培养基中,37℃振荡培养过夜;
2)按1:
100的比例将TG1过夜培养物接种到500mLSOB培养基中,37℃振荡培养至OD600为0.4;
3)将培养物冰浴30min,4℃,550g离心10min,沉淀细胞;
4)弃上清,用500mL预冷的无菌水水,4℃,550g离心10min,沉淀细胞;
5)弃上清用250mL预冷的无菌10%甘油溶液重悬细胞,4℃,600g离心10min,沉淀细胞;
6)用15mL预冷的无菌10%甘油溶液重悬细胞,4℃、600g离心10min,沉淀细胞;
7)将细胞悬浮在1mL预冷的CYT溶液中,将菌液按50µL/管分装在预冷的0.5mL离心管中,封紧管口,迅速没入液氮中冻存,保存于-70℃。
4.2.5电转化
1)将50µL感受态细胞从冰箱取出,冰上融化,加入2µL连接产物,混匀,冰上放置1min;(将所有的连接产物分多次进行电转);
2)将上述混合液转入0.2cm的电击杯中,调节电击参数:
电压为2.5kv,电场强度:
12.5kv/cm;
3)立即在电击杯中加入1mLSOC培养基,悬浮细胞,37℃培养1h;
4)取5µL适当梯度稀释,每个梯度分别取200µL涂布两块SOB-AG平板,37℃培养过夜,计算两块平板上的菌落数,用于计算库容;
库容=菌落总数/2×稀释倍数×菌液总体积(mL)/0.2×103
5)将剩余的培养物均分涂布平板,使得每块平板上的单菌落的疏密程度合适(不重合,但又不至于太稀),37℃培养过夜;
6)从平板上挑取一些转化子,进行PCR验证,分析插入率。
目的基因插入率=插入GB2的阳性克隆数/挑取的转化子总数×100%
实际库容=库容×目的基因插入率
7)每块平板上加入1mL2×YT培养基,用灭菌的药匙将细菌洗脱,将所有平板的细菌悬液合并,混匀。
取5mL细菌悬液用于噬菌体挽救,剩余的菌液则制备成20%的甘油保存液,–70℃冻存,此即为GB2随机突变大肠杆菌文库。
4.2.6辅助噬菌体滴度的测定
1)接种TG1于5mL2×YT培养基中,摇床培养至对数期后置于4℃预冷。
(可保存一周)
2)将试管中的3mL上层琼脂融化,将其温度平衡至48℃,每测一个滴度准备一管。
3)用2×YT培养基对待测辅助噬菌体噬菌体进行10倍梯度稀释。
4)将预冷的TG1培养物分装于1.5mL离心管中,每管100µL,每个稀释梯度一管。
5)将100µL不同稀释度的噬菌体分别加入到TG1培养物中,快速振荡混匀,37℃温育5min。
6)将温育后的混合物加入至上层琼脂中,迅速混匀后倾倒在2×YT平板上,待培养基凝结后倒置平板,37℃培养过夜,计算平板上的噬菌斑数量,以噬菌斑为30-300个左右的平板计数为准,计算噬菌体溶液的滴度。
4.2.7M13K07辅助噬菌体的扩增
1)从噬菌体滴度测定平板上,用牙签挑取一个独立的、噬菌斑较大的噬菌斑接种到5mL2×YT-K试管中,37℃、250rpm培养12h。
2)取500µL培养物接种到装有50mL2×YT-K培养基的三角瓶中,30℃、250rpm培养过夜。
3)收集过夜培养物,10000rpm离心20min,收集上清,含有噬菌体。
4)加入1/5体积的PEG/NaCl,充分混匀,让噬菌体4℃沉淀至少60min,最好过夜;
5)10000rpm离心20min,去上清,收集沉淀,溶解于8mL无菌PBS中。
6)测定M13K07溶液的滴度。
4.2.8GB2随机突变噬菌体文库的构建
1)将5mL细菌悬液转移至一50mL的离心管中,用2×YT培养基调整OD600至0.3,记录最终体积。
加入Amp使其终浓度为100µg/mL,加入无菌葡萄糖使其终浓度为2%,37℃振荡培养1h;
2)按phage:
cell=20:
1加入辅助噬菌体,37℃振荡培养2h;
3)1000g离心10min,弃上清,用50mL2×YT-AK培养基轻轻悬浮细胞,30℃振荡培养过夜;
4)收集培养物,10000g离心20min,将上清转移至10mL离心管中,上清中含有重组噬菌体;
5)加入1/5体积的PEG/NaCl,充分混匀,让噬菌体4℃沉淀至少60min,最好过夜;
6)4℃、1000g离心20min,倒掉上清,再次短暂离心,吸去残留的上清;
7)加入2mLPBS-1%BSA溶液重悬细胞,悬液转入10mL离心管中,4℃离心5min,沉淀残余的细胞;吸取上清,为并分装于1.5mL离心管中,加入50%甘油,-70℃保存备用。
此即为原始的GB2随机突变噬菌体文库;
8)测定文库滴度。
4.2.9噬菌体展示文库滴度的测定及无菌实验
1)接种TG1于5mL2×YT培养基中,摇床培养至对数期;
2)用SOB培养基10倍梯度稀释待测噬菌体文库;
3)待TG1培养物达到对数中期时,将其200µL等分于1.5mL离心管中,每个稀释梯度一管;
4)每管加入10µL的不同稀释度的噬菌体,快速振荡混匀,37℃温育10min,
5)将孵育后产物涂布于SOB-AG平板上,37℃倒置培养过夜。
6)计算平板上的单菌落数,计算噬菌体文库的滴度;
7)取10µL噬菌体涂布平板,37℃培养过夜,看是否有菌落生长。
4.2.10筛选靶分子鼠IgG的纯化
1)收集1mL小鼠腹水,4℃10000rpm离心10min,除去沉淀,将上清用0.45µm滤膜过滤,除去不溶性的小颗粒。
2)取1.5mLProteinGSepharose加入到层析柱中,用10倍体积的PBS进行过柱洗涤,注意液面,加上塞子,防止树脂暴露于空气中干燥。
3)加入0.5mL过滤后的小鼠腹水与0.5mLPBS混匀,盖上盖、塞后室温轻轻混匀15min,然后除去盖、塞,让液体流出。
4)加入10倍体积的PBS洗涤层析柱中树脂。
5)加入1mL0.1MGly-HCl(pH2.5)洗脱缓冲液,过柱洗脱,收集洗脱液,迅速加入适量的1MTris-HCl(pH9.0)中和缓冲液进行中和。
6)将获得的IgG洗脱液进行SDS-PAGE分析其纯度和浓度。
4.2.11IgG结合力减弱文库A的淘选
1)用PBS稀释IgG至终浓度为5μg/mL,按100µL/孔加入酶标孔中,4℃包被过夜。
2)PBST洗涤3次,加入2%BSA37℃封闭2h。
3)PBST洗涤3次,加入100µLPHEN-mtGB2噬菌体展示文库,噬菌体数量为1011cfu,37℃孵育1.5h。
4)用pH6.0的PBS洗涤缓冲液洗涤3次,然后加入300µL该缓冲液,37℃12min。
5)用pH6.0的PBS洗涤缓冲液洗涤10次,加入200µLpH4.0的Gly-HCl洗脱缓冲液,37℃孵育25min,将该洗脱缓冲液转移至一无菌离心管中,用中和缓冲液中和。
6)取10µL进行梯度稀释,测定滴度,计算淘选回收率,其余则加入至1mLTG1对数期培养物进行感染,37℃孵育1h,保存备用进行文库的扩增。
7)将文库扩增结果进行下一轮淘选,改变淘选条件,每一轮淘选条件如表4-2。
表4-2亲和力减弱文库A(pH4.0-6.0)各轮的淘选条件
Tab.4-2ThepanningconditionofeachroundsofthelibraryA(pH4.0-6.0)
轮次
IgG包被量(µg)
文库投入量(cfu)
洗涤时间(min)
洗脱时间(min)
1
0.5
1.0×1011
12
25
2
0.25
1.0×1011
15
25
3
0.1
2.0×1010
20
20
4
0.1
1.0×1010
20
15
4.2.12IgG结合增强文库B的淘选
1)用PBS稀释IgG至终浓度为5µg/mL,按100µL/孔加入酶标孔中,4℃包被过夜。
2)PBST洗涤3次,加入2%BSA37℃封闭2h。
3)PBST洗涤3次,加入100µLpHEN-mtGB2噬菌体展示文库,噬菌体数量为1011cfu,37℃孵育1.5h。
4)用pH7.0的PBST洗涤缓冲液洗涤10次。
5)用0.1MGly-HCl(pH2.4)洗涤缓冲液洗涤3次,加入300µL该缓冲液,37℃孵育12min,再用0.1MGly-HCl(pH2.4)洗涤缓冲液洗涤3次,然后用pH7.0的PBST洗涤缓冲液洗涤3次。
6)加入200µLTG1对数期培养物,37℃感染1h。
7)取出感染后TG1培养物,取10µL进行梯度稀释,测定滴度,计算淘选回收率,其余则进行文库的扩增。
8)将扩增后文库进行下一轮淘选,改变淘选条件,每一轮淘选条件如表4-3。
表4-3亲和力减弱文库B(pH<pH2.4)各轮的淘选条件
Tab.4-3ThepanningconditionofeachroundsofthelibraryB(pH<pH2.4)
轮次
IgG包被量(µg)
文库投入量(cfu)
洗涤时间(min)
1
0.5
1.0×1011
12
2
0.25
2.0×1010
15
3
0.1
5.0×109
20
4
0.1
5.0×109
25
4.2.13淘选后文库的扩增
1)将文库感染物产物加入至3mL2×YT-G培养基中,37℃、225rpm振荡培养1h,加入Amp至终浓度为100ng/mL,同时按cell:
phage=1:
20的比例加入M13K07噬菌体,37℃、225rpm振荡培养2h。
2)将培养物分装于离心管中,1000rpm,5min,收集菌体,加入至20mL2×YT-AK培养液中,30℃,250rpm振荡培养过夜。
3)将过夜培养物10000rpm离心20min,收集上清,每5mL上清中加入1mLPEG/NaCl,混匀后置于4℃1h以上,最好是过夜。
4)10000rpm,20min,去除上清,将沉淀重悬于2mLTBS中,加入1/5体积的PEG/NaCl,混匀后置于4℃1h以上。
5)10000rpm,20min,去除上清,将沉淀悬浮于400µLTBS-1%BSA中,即为该轮淘选后文库,测定滴度,用于下一轮淘选或者分析。
4.3实验结果
4.3.1野生型pHENI-GB2质粒的构建
通过NcoI和XhoI将野生型wtGB2克隆至pHENI中,菌落PCR验证后提取阳性克隆子的质粒,用NcoI和XhoI进行双酶切验证,结果获得两条带,分别与空载体和外源片段大小一致,说明载体构建成功(图4-4)。
进一步经测序确正,未发现突变。
图4-4pHENI-wtGB2重组质粒的NcoI和XhoI双酶切验证图
Fig.4-4IdentificationofthepHENI-wtGB2byrestrictionenzymeofNcoIandXhoI
M1:
λ-HindⅢdigestDNAmarker;M2:
DL2000marker;
1:
pHENI噬菌粒双酶切样品;2:
pHENI-wtGB2重组噬菌粒双酶切样品;
3:
mtGB2DNA片段双酶切样品
4.3.2GB2的随机突变
以pHENI-wtGB2重组噬菌粒为模板,pHEN-F和pHEN-R为引物,采用易错PCR方法对GB2进行随机突变,分四管进行,四管分别将一种脱氧核苷酸的浓度降低90%,每个反应都进行35个循环。
1.0%琼脂糖凝胶电泳分析,四个反应均成功进行,产物浓度、大小也一致(见图4-5)。
图4-5易错PCR扩增产物的琼脂糖凝胶电泳图
Fig.4-5Analysisoftheerror-PCRproducts
M:
DL2000Marker;1、2、3、4:
分别为减少90%的dATP、dTTP、dCTP和dGTP的易错PCR扩增产物;
将4管易错PCR产物混合,经乙醇沉淀浓缩纯化,取部分进行TA克隆,经菌落PCR验证后,挑取4个克隆子送测序,用DNASTAR软件将其与野生型GB2片段进行比较,分析随机突变情况。
由图4-6可以看出共发生了29处点突变,突变率约为1.93%。
随机突变位点的分布位置是随机的,没有出现突变热点。
就突变碱基而言,主要是A、T发生突变,占了约82%,突变结果表现出了一定的偏向性。
图4-6随机突变产物的氨基酸突变结果分析图
Fig.4-6ComparisonoftheaminoacidsequencesbetweenwtGB2andfourmtGB2clonesfromerror-PCR
黄色:
发生点突变;红色:
未发生点突变


- 配套讲稿:
如PPT文件的首页显示word图标,表示该PPT已包含配套word讲稿。双击word图标可打开word文档。
- 特殊限制:
部分文档作品中含有的国旗、国徽等图片,仅作为作品整体效果示例展示,禁止商用。设计者仅对作品中独创性部分享有著作权。
- 关 键 词:
- GB2随机突变噬菌体文库的构建及淘选 GB2 随机 突变 噬菌体 文库 构建 淘选
 冰豆网所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
冰豆网所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
 《初级会计实务》试题题库大全及答案详解.docx
《初级会计实务》试题题库大全及答案详解.docx
-
《管理学》习题教材15章.docx
-
《教育学》读后感范文精选6篇.docx
-
《林教头风雪山神庙》练习题.docx
-
《企业文化》期末复习应考指南央专.docx
-
《数据结构》知识题汇编09第九章排序试题.docx
-
《偷影子的人》读后感集合15篇.docx
-
《幼儿园工作规程》.docx
-
《残疾人证》管理办法.docx
-
《故事》教学反思.docx
-
《Java语言学习知识程序设计》复习资料汇编.docx
-
《短文两篇》课堂实录.docx
-
《基于MATLAB的信号与系统实验指导》编程练习试题doc.docx
-
《昆虫记》好词好句大全.docx
-
《木棉树》阅读答案.docx
-
《区间信号自动控制》练习册答案.docx
-
《山东省中小学教师职称评审表》高级教师一级教师二级教师专用A4纸正反面打印按页码装订许知忠.docx
-
《安娜卡列尼娜》读后感.docx
-
《繁星春水》读后感15篇.docx
-
《苏州市市级示范物业管理项目服务质量评价标准》 doc.docx
-
《采薇》教案.docx
-
《假如给我三天光明》阅读测试题有答案.docx
-
《小学数学教师》读书笔记精选多篇.docx
-
《给幼儿教师的一把钥匙》读书笔记.docx
-
《劳动法》教案设计.docx
-
《综合基础知识》必看考点《刑法》含答案.docx
-
《建筑构造》考试试题及答案精华.docx
-
3套打包北师大版四年级下册英语期末单元测试题解析版.docx
-
《雷锋的微笑》观后感.docx
-
《女人故事》电视栏目策划方案1.docx
-
7万多车对比分解.docx
-
《调皮的日子》题库.docx
-
西师版小学三年级上册语文全册教案及积累运用.docx
-
新大象版科学五年级下册全册教学设计精品docx.docx
-
新人教版一年级数学上册第八单元20以内的进位加法教案.docx
-
学年高中物理必修一第二章 学案6自由落体.docx
-
学习普通话的体会.docx
-
英语高中英语代词技巧和方法完整版及练习题.docx
-
中考初中语法讲解定语从句.docx
-
最新年度社区居委会工作总结范文工作总结文档五篇.docx
-
《艾青诗集》名篇摘抄赏析.docx
-
《电子设计大赛》word版.docx
-
《会计岗位实训》习题标准答案.docx
-
《念奴娇赤壁怀古》教案.docx
-
教学设计原理加涅完整笔记.docx
-
特种设备作业人员考试中心质保手册.docx
-
教育局教育质量优化工作半年总结与教育局教育资源统筹发展半年工作总结汇编.docx
-
天津一中高三理科综合能力测试第五套.docx
-
大班健康教育好玩的皮筋教案.docx
-
通宝安全生产管理办法讨论稿.docx
-
道路及铺装施工方案.docx
