 分子生物学实验指导dodocWord下载.docx
分子生物学实验指导dodocWord下载.docx
- 文档编号:19388848
- 上传时间:2023-01-05
- 格式:DOCX
- 页数:22
- 大小:74.34KB
分子生物学实验指导dodocWord下载.docx
《分子生物学实验指导dodocWord下载.docx》由会员分享,可在线阅读,更多相关《分子生物学实验指导dodocWord下载.docx(22页珍藏版)》请在冰豆网上搜索。
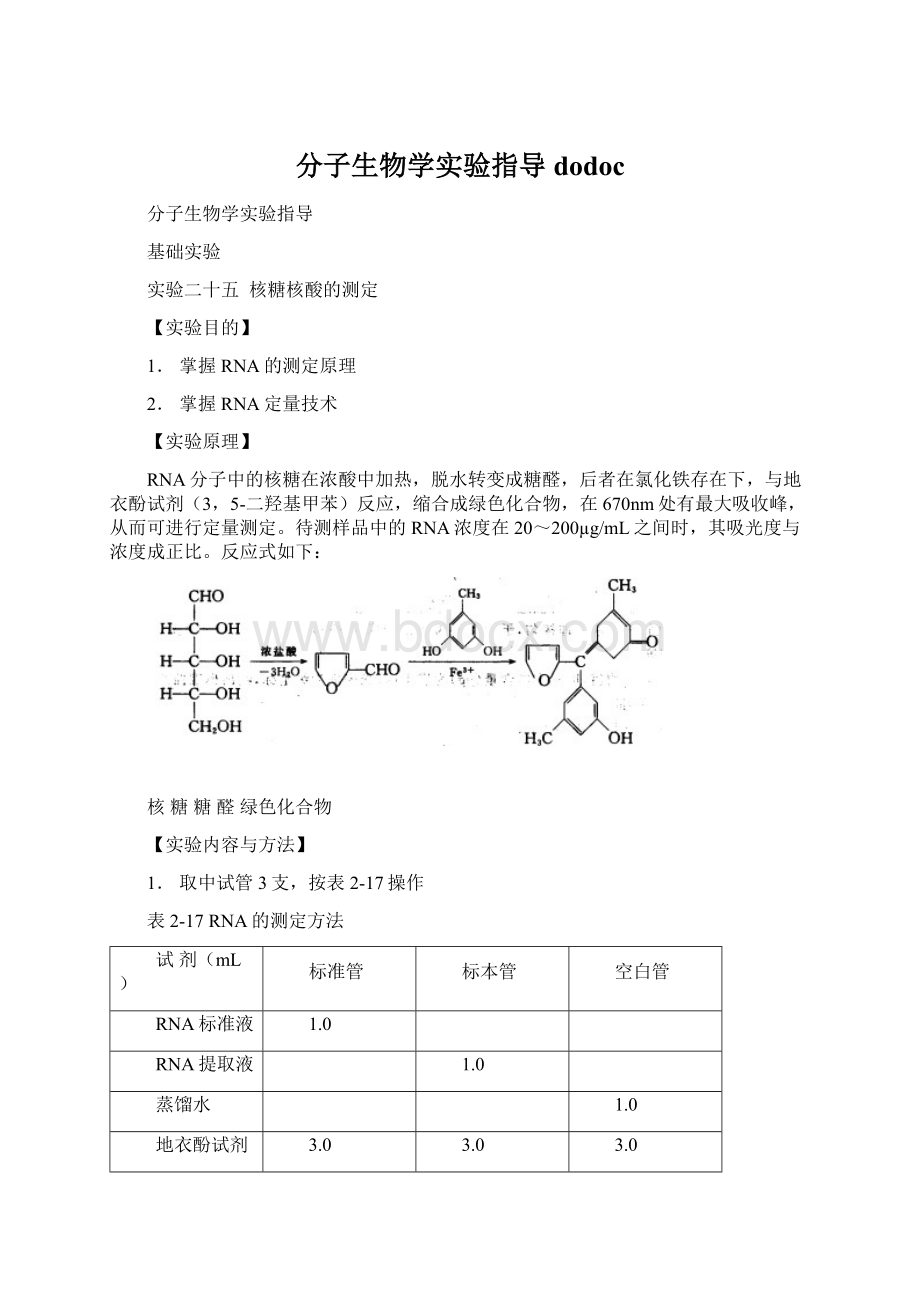
【实验准备】
1.RNA标准液准确称取RNA用0.001mol/LNaOH配制成100mg/mL的溶液。
2.地衣酚试剂称取地衣酚(3,5-二羟基甲苯)100mg,溶于100mL浓盐酸(AR)中,再加入100mgFeCl3.6H2O。
应用前新鲜配制。
3.仪器:
紫外可见分光光度计、试管、刻度吸管
(孔峰张孟业)
实验二十六脱氧核糖核酸提取
掌握DNA分离纯化的原理和方法
见RNA提取。
1.肝匀浆制备与RNA分离,见RNA的提取。
2.于含有DNP的沉淀中加入二倍体积的1mol/LNaCl溶液搅拌均匀,室温放置5~6h,以充分提取DNP。
3.去除蛋白质将放置完毕的DNP混悬液离心3000rpm/10min,上层转移至另一试管中,加入等体积氯仿-异戊醇混合液,用玻璃纸堵住管口振摇2min,离心3000rpm/10min,此时管中液体分为三层,下层为氯仿,中层为变性的蛋白质,上层为含有DNA的水溶液,将上层转移至另一小试管中(中、下层弃去),加二倍体积冰冷的95%乙醇,边加边摇匀,此时可见有白色纤维状DNA(或为乳白色紊状沉淀),离心3000rpm/5min,弃去上层乙醇溶液,沉淀即为DNA的粗制品。
1.1.0mol/LNaCl溶液
2.仪器:
高速组织捣碎机、离心机、紫外可见分光光度计
【注意事项】
(1)在制备肝匀浆时,应尽量在冰冷条件下进行,并尽快加入0.14mol/L氯化钠溶液,以抑制脱氧核糖核酸酶,防止DNA的分解以提高核酸得量。
(2)各步骤操作应力求准确,尽量减少中途核酸的丢失,保证定量测定的准确性。
实验二十七脱氧核糖核酸的测定
1.掌握DNA的比色测定原理
2.掌握DNA定量技术
DNA分子在酸性溶液中加热水解后,其脱氧核糖部分可被浓酸缩水成ω-羟基γ-酮基戊醛等化合物,再进一步与二苯胺作用,可产生蓝色化合物,在595nm处有最大吸收峰。
从而可进行定量测定。
待测样品中的DNA浓度在20~200µ
g/mL之间时,其吸光度与浓度成比。
脱氧核糖ω-羟基-γ-酮基戊醛
1.取中试管3支,编号,按表2-18操作
表2-18DNA的测定方法
DNA标准液
DNA提取液
二苯胺试剂
2.0
2.混匀各管,置于沸水浴中加热10min,取出冷却,以空白管调零点,在595nm处比色,读取各管的吸光度。
3.计算:
1.DNA标准液准确称取DNA用0.01mol/LNaOH配制成400µ
g/mL溶液。
2.二苯胺试剂称取二苯胺1.5g(如不纯,须用70%乙醇重结晶两次),溶于100mL冰醋酸(AR)中,再加入浓硫酸1.5mL,贮存在棕色瓶中放入冰箱内保存备用。
紫外可见分光光度计,试管,刻度吸管。
【思考题】
1.本实验中,测定RNA和DNA含量的原理是什么?
2.核酸在细胞质和细胞核中的分布有何特点?
实验二十八真核细胞DNA的制备与定量
掌握真核生物细胞基因组DNA制备和定量的基本方法。
制备基因组DNA是进行基因结构和功能研究的重要步骤,通常要求得到的片段长度不小于100~200kb。
在DNA提取过程中应尽量避免使DNA断裂和降解的各种因素,以保证DNA的完整性。
一般真核细胞基因组DNA有107~109bp,可以从新鲜组织、培养细胞或低温保存的组织细胞中提取。
在EDTA以及SDS等试剂存在下用蛋白酶K消化细胞,随后用酚抽提而获得DNA,DNA经酶切后可用于Southern分析,还可用于PCR的模板、文库构建等实验。
根据材料来源不同,采取不同的材料处理方法,而后的DNA提取方法大体类似,但都应考虑以下两个原则:
①防止和抑制DNase对DNA的降解;
②尽量减少对溶液中DNA的机械剪切破坏。
1.细胞处理
(1)收集对数生长期的培养细胞约5×
106于1.5mL离心管中,离心1000rpm/5min,弃上清液。
(2)加0.01mol/L的PBS(pH7.2)缓冲液1mL重悬细胞,离心1000rpm/5min,弃上清液。
(3)于振荡器上打散细胞,加500μL裂解缓冲液,充分混匀后置于50℃~55℃水浴1~2h。
2.DNA提取
(1)加等体积Tris饱和酚至上述样品处理液中,温和、充分地混匀,4℃离心12000rpm/5min。
(2)小心吸取上层水相到另一1.5mL离心管中,加等体积酚/氯仿,轻轻混匀,4℃离心12000rpm/5min。
(3)取上层水相到另一1.5mL离心管中,加等体积氯仿/异戊醇,轻轻混匀,4℃离心12000rpm/5min。
(4)转移上层水相到另一1.5mL离心管中,加1/10体积的3mol/L醋酸纳(pH5.2)和2.5倍体积的无水乙醇,轻轻颠倒混匀。
(5)待絮状物出现后,4℃离心12000rpm/5min。
(6)弃上清液,沉淀用75%乙醇洗涤,离心12000rpm/2min。
(7)弃上清液,室温下挥发乙醇,待沉淀将近透明时加50~100μlTE置4℃冰箱溶解过夜。
3.DNA定量:
DNA在260nm处有最大的吸收峰,蛋白质在280m处有最大的吸收峰,盐和小分子则集中在230m处。
因此,可以在260nm波长处测定DNA吸光度,OD260值为1时约相当于50μg/ml的双链DNA。
取1µ
l所得DNA,加49µ
l双蒸水,于260nm处测定其吸光值。
4.结果分析:
根据OD260值可计算出DNA样品的浓度:
DNA(mg/ml)=50×
OD260×
稀释倍数/1000。
DNA纯品的OD260/OD280为1.8,故根据OD260/OD280的值可以估计DNA的纯度。
若比值较高,说明含有RNA,比值较低说明有残余蛋白质存在。
1.TE缓冲液10mmol/LTris-HCl(pH7.8)、lmmol/LEDTA(pH8.0)
2.0.01mol/L的PBS(pH7.2)缓冲液
3.裂解缓冲液10mmol/LTris-HCl(pH8.0),15mmol/LNaCl,10mmol/LEDTA(pH8.0),1%SDS。
使用前加入蛋白酶K至100μg/ml。
4.Tris饱和酚(pH8.0)
5.酚/氯仿(1/1)
6.氯仿/异戊醇(24/1)
7.无水乙醇、75%乙醇
1.所有用品均需要高温高压,以灭活残余的DNA酶。
2.所有试剂均用高压灭菌双蒸水配制。
3.用大口滴管或吸头操作,以尽量减少打断DNA的可能性。
1.制备真核生物细胞基因组DNA时需注意什么?
2.如何检测DNA?
(孔峰田克立)
实验二十九质粒DNA的提取与定量
掌握碱裂解法小量制备质粒DNA的方法。
分离质粒DNA的方法是依据其与染色体DNA分子大小不同、碱基组成的差异以及质粒DNA的超螺旋共价闭环状结构的特点来进行。
目前常用的提取质粒的方法有碱裂解法、煮沸法和去污剂法等。
本实验采用的是碱裂解法。
其原理是将细菌菌膜破裂后,利用质粒DNA与染色体DNA之间变性与复性的差异而达到分离的目的,因为①染色体DNA分子量比质粒DNA大得多;
②从细胞中提取到的染色体DNA大多断裂成线状分子,而质粒DNA则为共价闭合环状结构。
在pH高达12.6的条件下,染色体DNA的氢键断裂,双螺旋结构解开变性,同时由于受到机械切力或核酸酶等的作用,染色体DNA易被切成大小不同的片段;
而闭环质粒DNA链氢键也断裂,但它的超螺旋共价闭合环状的两条互补链不会完全分离,不易发生变性。
当以pH4.8的醋酸钾高盐缓冲液调节pH至中性时,质粒DNA又恢复其天然构象,以可溶状态存在于液相中,而染色体DNA不能复性而形成缠连的不溶性网状结构,与不稳定的大分子RNA、变性的蛋白质-SDS复合物以及细菌碎片等一起沉淀而被除去。
再进一步用酚、氯仿使蛋白质变性去除蛋白杂质,然后用无水乙醇沉淀质粒DNA,可得到纯化的质粒DMA。
1.操作流程:
挑取单菌落接种到含amp的LB液体培养基管内(3.5ml/管)
↓放37℃培养箱内振荡培养过夜
将菌液倒入1.5ml离心管中(尽量倒满)12000rpm/30s(沉淀菌体)
↓在原管中重复上述操作1次
弃上清扣干,加入预冷的solutionⅠ100μL,剧烈振荡打散菌体,冰浴5min
↓(以下操作要轻柔)
加入新配制的solutionⅡ200μL,温和颠倒离心管2~3次混匀,室温放置5min
↓(此时液体应由混浊变为透明粘稠)
加入预冷的solutionⅢ150μL,温和颠倒离心管2~3次混匀,冰浴5min
↓离心12000rpm/5min
小心将含有质粒DNA的上清移至另一离心管中
↓
加等体积氯仿,轻微振荡,离心12000rpm/2min
↓转移上清液至另一离心管
加入2倍体积冰冷无水乙醇,轻轻混匀室温放置2分钟
↓离心12000rpm/5min
弃上清,加入70%乙醇洗涤沉淀,离心12000rpm/2min,弃上清
↓重复洗涤1次
弃上清,液体尽量倒净并在吸水纸上扣干
↓室温放置15min干燥
加入1×
TE30μL溶解质粒,用于定量分析、电泳检测等。
1.氨苄青霉素(amp)将amp配制成200μg/μl溶液,分装小管,-20℃保存。
2.LB液体培养基取胰蛋白胨2g,酵母提取物1g,氯化钠1g,加水160ml溶解后用1mol/L氢氧化钠调pH至7.0,定容至200ml,高压灭菌。
临用前加200μg/μl的amp60μl(每mlLB含amp60μg)。
3.固体培养基取胰蛋白胨2g,酵母提取物1g,氯化钠1g,琼脂粉3g,加水160ml溶解后用1mol/L氢氧化钠调pH至7.0,定容至200ml,高压灭菌。
临用前加200μg/μlamp60μl,将固体培养基倒入平皿中,待凝固后备用。
4.SolutionⅠ取蔗糖17.13g、Tris3.03g、EDTA3.72g,加双蒸水定容至500mL后8磅/英寸2高压灭菌,或过滤除菌,4℃贮存。
5.SolutionⅡ0.2mol/L氢氧化钠,1%SDS,临用前新鲜配制。
注意:
SDS易产生气泡,不要剧烈搅拌。
6.SolutionⅢ5mol/LKAc60mL,冰醋酸11.5ml,加双蒸水定容至100ml,高压灭菌,4℃贮存。
1.质粒DNA提取过程中动作要轻柔,以免对DNA造成损伤;
2.加入solutionⅠ后一定要充分打散菌体;
3.加入solutionII后放置时间不要超过5min,以免变性质粒不易复性。
4.加入solutionII后液体应由混浊变为透明粘稠,可见拉丝现象。
若没有上述现象发生,则实验不能继续下去,应检查试剂配制及操作是否有误,然后重新开始。
1.简述碱裂解法提取质粒的基本原理。
2.提取质粒DNA应注意哪些问题?
实验三十DNA琼脂糖凝胶电泳
掌握琼脂糖凝胶电泳的原理和基本操作技术。
DNA的电泳原理与蛋白质电泳基本相同。
在pH高于其pI时,DNA分子带负电荷,在直流电场中DNA向正极泳动。
不同的DNA分子因所带电荷、构象和分子量大小不同,在同一电泳系统中的泳动速度存在差异,因而可以使核酸片段彼此分离,并检测其分子大小。
电泳后的凝胶浸泡于荧光染料(如溴化乙锭)中,染料分子可嵌入DNA分子碱基之间,在紫外线照射下发出荧光,可观察到核酸片段所在的位置和亮度,从而对DNA进行定性或定量测定。
琼脂糖凝胶的浓度可根据DNA分子的大小来确定,一般基因组DNA可用0.8%琼脂糖凝胶进行电泳检测,质粒DNA选择1%琼脂糖凝胶,而对只有几百个碱基对的寡核苷酸可制备浓度更高一些的琼脂糖凝胶。
1.凝胶板的制备取有机玻璃胶槽,洗净、晾干、安装好(不同型号的电泳槽有不同的安装方法),水平放置,注意防止漏胶。
将梳子插入到胶槽的定位槽中。
2.灌胶:
将熔化的琼脂糖凝胶冷却至约65℃时,小心地将凝胶倒入胶槽上,控制灌胶速度,使胶缓慢地展开,直到整个有机玻璃板表面形成均匀的凝胶层,凝胶厚度一般为0.3~0.5cm。
3.室温下放置约1h,待胶凝固完全后,将铺胶的有机玻璃胶槽放在电泳槽中,加入0.5×
TBE电泳缓冲液,液面高于凝胶面约0.5cm,轻轻拔出梳子。
4.加样将实验二十八和实验二十九所得的基因组DNA和质粒DNA作为上样样品。
将样品和上样缓冲液6:
1混匀,用微量加样器将样品分别加入胶板的样品小槽内(注意:
加样时应防止加样器头碰坏凝胶孔壁)。
5.电泳加样完毕后将靠近样品槽一端连接负极,另一端连接正极,接通电源,开始电泳,为防止样品扩散,在样品进胶前可用略高电压。
当样品进胶后,应控制电压不高于5V/cm。
当染料条带移动到距离凝胶前沿1cm时,停止电泳。
6.染色将电泳后的胶板小心推进含溴化乙锭(0.5μg/mL)的染色液中,室温下浸泡约30min。
7.观察小心取出凝胶并用水轻轻冲洗胶表面的溴化乙锭溶液,将凝胶放在紫外灯观察台上,在波长254nm紫外灯下进行观察。
8.结果分析:
在DNA存在的位置呈现橘红色荧光,肉眼可观察到清晰的条带。
基因组DNA分子量很大,一般在点样孔附近有单一的高分子量条带。
质粒DNA由于不同构型的DNA在琼脂糖凝胶电泳中的迁移率不同,因此在琼脂糖凝胶电泳可出现三条电泳带,分别为三种构型的质粒DNA,SC最快,LC次之,OC最慢。
1.5×
TBE取Tris5.4g,,硼酸2.75g,EDTA-Na20.37g,用蒸馏水定容至1000ml。
2.称取制备0.8%和1.0%凝胶的琼脂糖,加0.5×
TBE微波炉中融化,略冷之后进行灌胶。
3.6×
上样缓冲液0.25%溴酚蓝,40%(w/v)蔗糖溶液,贮存于4℃。
4.溴化乙锭配制成10mg/ml,用铝箔或黑纸包裹容器,室温储存。
1.EB是强诱变剂并有中等毒性,配制和使用时都应戴手套,并且不要把EB洒到桌面或地面上。
凡是沾污了EB的容器或物品必须经专门处理后才能清洗或丢弃。
2.当EB太浓,胶染色过深,DNA带看不清时,可将胶放入蒸馏水冲泡30分钟后再观察。
1.琼脂糖凝胶电泳中DNA分子迁移率受哪些因素的影响?
2.简述EB染色的原理及实验注意事项。
实验三十一DNA的限制性酶切反应
掌握DNA限制性内切酶酶切的基本原理。
限制性内切酶能特异地识别、结合于一段被称为限制性酶序列的DNA序列之内或其附近的特异位点上,并切割双链DNA。
它可分为三类,其中Ⅱ类限制性内切酶在分子克隆中得到了广泛应用,它们是重组DNA技术的基本工具酶。
绝大多数Ⅱ类限制酶可识别长度为4至6个核苷酸的回文序列,其切割位点在识别序列中,有的在对称轴处切割双链DNA,产生平末端的DNA片段;
有的切割位点在对称轴一侧,产生带有单链突出末端的DNA片段称粘性未端。
1.用HindⅢ酶切:
提取的质粒(pUC18或pBR322)30μL
加入10×
MBuffer4μL,双蒸水4μL,加HindⅢ2μL(12U/μL)
↓(终体积为40μL)
37℃水浴2h,用电泳检查酶切结果。
2.用PstⅠ酶切
向上述酶切体系中加入1/10体积(4μL)的3mol/LNaAC溶液混匀
加入2倍体积的无水乙醇,室温放置5min
↓离心12000rpm/5min
去上清,向沉淀(DNA)中加入70%乙醇洗涤倒净液体,扣干
↓(注:
如果沉淀被冲开需再离心,12000rpm,2min,70%乙醇洗涤,可重复2~3次)
室温放置15~30min,自然干燥
↓加10μL1×
TE溶液溶解DNA
加入10×
Hbuffer2μL,双蒸水6μL,PstⅠ2μL(12U/μL)
↓(终体积为20μL)
37℃水浴2h,用电泳检查酶切结果
3.结果分析:
酶切样品与DNA分子量标准共同进行琼脂糖凝胶电泳,电泳后在相当于质粒DNA分子量大小的位置显示肉眼可辨的荧光条带(EB染色)。
1.pUC18(或pBR322)质粒
2.HindⅢ酶及其酶切缓冲液
3.PstI酶及其酶切缓冲液
(1)限制性内切酶需保存于-20℃,操作时应将酶保持在冰浴中,避免长时间置于高温中。
(2)在酶切反应中,应最后加入酶,尽量减少室温接触机会。
(3)加样时吸头垂直进入试剂管,避免碰到管壁,加酶时吸头深入不可过深。
每加完一样要换一个吸头,同时在已加的样品前做个记号以防止错加或漏加,避免污染。
(4)注意酶的用量,加入的酶量按1-3U/ugDNA计算,酶的体积应低于反应总体积的10%,以避免酶液中甘油干扰反应。
酶量过大时,有产生星号活性的可能,即在识别序列以外的位点进行切割。
(5)酶解消化反应温度及时间根据该酶使用说明书而定。
1.酶切时应注意什么?
2.如果一个DNA酶解液在电泳后发现DNA未被切动,可能是什么原因?
实验三十二目的DNA片段的回收
掌握从琼脂糖凝胶中回收纯化DNA片段的方法。
分离和纯化DNA酶切片段是基因工程中常用的手段。
在构建重组DNA分子时,为了提高重组效率,载体DNA和目的基因的酶切片段、包括化学合成的基因、PCR扩增的产物,以及DNA标记反应前后的DNA片段等都需要进一步进行分离纯化与回收。
另外在检测重组DNA的分子杂交技术中,要制备DNA探针,往往也要先分离回收以获得纯度较高的DNA片段。
目前用于回收DNA片段的方法很多,可以根据待回收DNA片段的纯度、大小、浓度和实验室现有的条件等选择合适的方法。
1.酶切之后进行琼脂糖凝胶电泳,电泳结束后在长波紫外灯下切出含有目的DNA片段的凝胶,注意去除多余凝胶,尽量减少胶体积(切胶时注意不要将DNA长时间暴露于紫外灯下,以防止DNA损伤)。
2.称量胶块重量,计算胶块体积:
1mg=1μl。
3.将胶块尽量切碎,以提高DNA的回收率。
4.向胶块中加入3倍凝胶体积量的胶块融化液DR-Ibuffer。
5.置75℃水浴中加热融化胶块,期间间断振荡混合,使胶块充分融化(约6~10min)。
6.向上述胶块融化液中加入DR-Ibuffer量的1/2的DR-IIbuffer,混合均匀。
7.将SpinColumn管安放于Collection管上。
8.将操作6中的溶液转移至SpinColumn中,离心3600rpm/1min,弃滤液。
(可将滤液再加入SpinColumn中离心一次,可提高DNA的回收率)
9.将500μl的RiuseA液加入SpinColumn中,离心3600rpm/30s,弃滤液。
10.将700μl的RiuseB液加入SpinColumn中,离心3600rpm,30s,弃滤液。
11.重复步骤10,然后再离心12000rpm/1min.
12.将SpinColumn放置于一只新的1.5ml离心管上,在SpinColumn膜中央出加入25μl水或TE,室温静置1min。
13.离心12000rpm,1min,洗脱回收DNA。
14.结果分析:
回收纯化的DNA经琼脂糖凝胶电泳检测可见特定分子量大小的单一的清晰条带。
1.1%琼脂糖凝胶
2.待纯化的DNA
3.琼脂糖凝胶DNA回收试剂盒
4.去离子水或TE(pH7.6)
紫外光对DNA分子有切割作用,从胶上回收DNA时,应尽量缩短光照时间并采用长波长紫外灯(300-360nm),以减少紫外光对DNA的损伤。
【思考题】从凝胶中回收DNA,切胶时需注意什么?
实验三十三大肠杆菌感受态细胞的制备
掌握制备大肠杆菌感受态细胞的方法。
体外连接的重组DNA分子导入合适的宿主细胞中才能大量的进行复制、增殖和表达。
细菌处于容易吸收外源DNA的状态叫感受态。
CaCl2法是目前实验室常用的制备感受态细胞的方法,其原理是使细菌处于0℃、CaCl2低渗溶液中,细胞膨胀成球形,细胞膜的通透性发生改变,外源DNA附着于细胞膜表面,经42℃短时间热冲击处理,促进细胞吸收DNA复合物。
本实验以JM109菌珠为受体细胞,用CaCl2处理受体菌使其处于感受态。
转化是指质粒DNA或以它为载体构建的重组子导入细菌的过程。
利用pBR322(或pUC18)质粒转化制备的感受态菌,可用于所制备感受态菌的转化鉴定。
pBR322质粒携带有氨苄青霉素和四环素的抗性基因,可使接受了该质粒的受体菌具有了氨苄青霉素和四环素抗性,将经过转化后的受体细胞经过适当稀释,在含氨苄青霉素和四环素的平板培养基上培养,只有转化子才能存活,而未受转化的受体细胞则因无抵抗氨苄青霉素和四环素的能力而死亡。
1.感受态细胞的制备
(1).从新活化的JM109菌平板上挑取单菌落,接种于5mlLB液体培养基中(不含Amp),37℃振荡培养12h左右,至对数生长期,将该菌液以1:
100~1:
50转接于100mlLB培养基中,37℃振荡培养2~3h(OD600约0.2~0.4)。
(2).将菌液冰浴10min后,转入离心管中,于4℃离心4000rpm,10min,去上清,收集菌体。
(3).用10ml冰预冷的0.1mol/LCaCl2悬浮细胞,冰浴15~30min。
(4).0~4℃,离心4000rpm,10min。
(5).弃上清液,每50ml初始培


- 配套讲稿:
如PPT文件的首页显示word图标,表示该PPT已包含配套word讲稿。双击word图标可打开word文档。
- 特殊限制:
部分文档作品中含有的国旗、国徽等图片,仅作为作品整体效果示例展示,禁止商用。设计者仅对作品中独创性部分享有著作权。
- 关 键 词:
- 分子生物学 实验 指导 dodoc
 冰豆网所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
冰豆网所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
 铝散热器项目年度预算报告.docx
铝散热器项目年度预算报告.docx
-
牛津上海版通用小学英语三年级上册Unit 12同步练习2II 卷.docx
-
论我国私营企业员工激励机制.docx
-
人教版五年级品德与社会上册全册教案.docx
-
开学啦国旗下讲话稿三分钟.docx
-
露天采矿学复习题.docx
-
六年级英语教师年度考核个人总结.docx
-
某路站综合体项PC吊装施工方案.docx
-
人教版九年级历史上册期末考试试题一套.docx
-
隆昌妇幼保健院.docx
-
芦二矿抽采达标中长期规划.docx
-
看拼音写词语.docx
-
模拟磁盘调度算法系统的设计毕业设计.docx
-
每周一条名言警句或一首诗词.docx
-
棉花膜下滴灌示范工程设计总结报告.docx
-
九年级化学教案第十单元酸和碱教案新人教版.docx
-
宁波市水资源公报.docx
-
农业实用技术培训工作意见与农业局上半年工作总结范例两篇汇编.docx
-
平行线的判定.docx
-
内部会计管理制度11成本核算制度.docx
-
盘扣式脚手架支撑方案.docx
-
旅游规划模板.docx
-
煤矿大本大专毕业设计大采高综采工作面作业规程.docx
-
美学选择题整理课件资料.docx
-
名家论腹泻慢性肠炎.docx
-
宁夏银川市第一中学学年高一上学期期中考试地理试题解析解析版.docx
-
年产吨精密纤维纸项目建设建议书.docx
-
农技推广中心工作总结.docx
-
彭宇案的法逻辑批判.docx
-
宁夏仕奇房产网发布份房地产交易情况.docx
-
项目推荐书智能温控节能系统.docx
-
区县节日期间加强消防安全讲话稿与区发改委领导班子述职述廉报告汇编.docx
-
大学各专业对应英文名称.docx
-
大学生考证情况分析.docx
-
大学生职业生涯规划书最新范文总结.docx
-
超市促销广播稿.docx
-
参观超市心得体会3篇.docx
-
仓库管理员个人总结1000字.docx
-
沉井专项施工方案.docx
-
诚信伴我成长主题班会方案与对策.docx
-
测量学实习报告 参考.docx
-
初中英语模拟试题.docx
-
城镇化进程中的农村社会管理问题研究以三亚市为例.docx
-
产品质量管理制度最新版.docx
-
传播学讲义1.docx
-
创新基金成功案例.docx
-
春节送给老师的祝福语短信.docx
-
初步设计专家意见.docx
-
常州教科研课题分析情况登记表.docx
-
从业资格考试备考《中药学专业知识二》考前复习题含答案解析第三十六篇.docx
-
初三化学复习课教案.docx
