 融合蛋白表达实验报告文档格式.docx
融合蛋白表达实验报告文档格式.docx
- 文档编号:18841804
- 上传时间:2023-01-01
- 格式:DOCX
- 页数:23
- 大小:651.50KB
融合蛋白表达实验报告文档格式.docx
《融合蛋白表达实验报告文档格式.docx》由会员分享,可在线阅读,更多相关《融合蛋白表达实验报告文档格式.docx(23页珍藏版)》请在冰豆网上搜索。
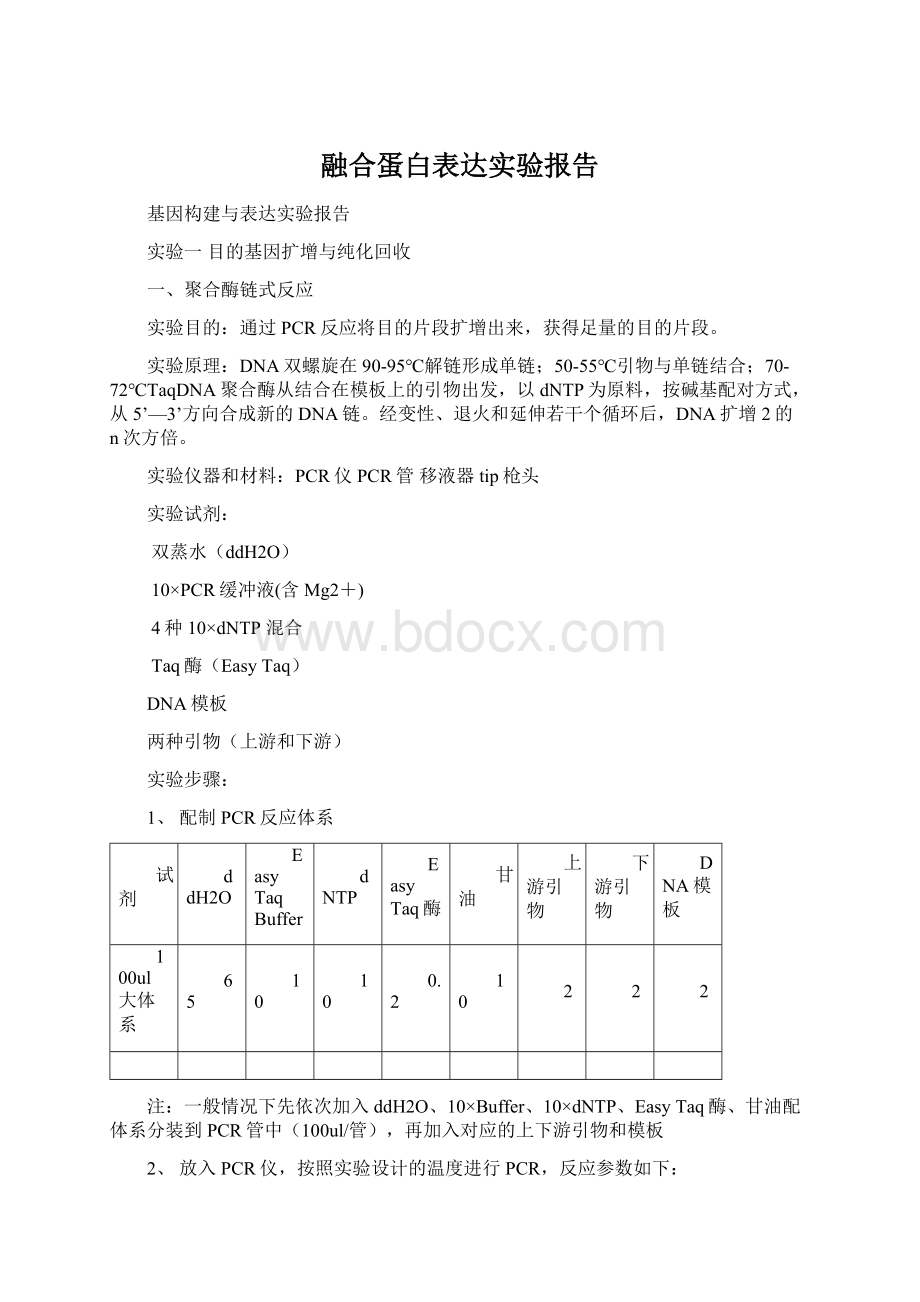
一般情况下先依次加入ddH2O、10×
Buffer、10×
dNTP、EasyTaq酶、甘油配体系分装到PCR管中(100ul/管),再加入对应的上下游引物和模板
2、放入PCR仪,按照实验设计的温度进行PCR,反应参数如下:
(1)94℃预变性4min;
(2)94℃变性1min;
(3)55℃退火1.5min;
(4)72℃延伸2min;
(5)重复
(2)-(4)30个循环;
(6)72℃终延伸10min;
二、琼脂糖凝胶电泳检测
通过琼脂糖凝胶电泳检测DNA分子量的大小,检测目的基因片段是否扩增出来。
DNA分子在琼脂糖凝胶中泳动时有电荷效应和分子筛效应,DNA在碱性溶液中带有负电荷,在电场作用下朝正极移动。
在琼脂糖凝胶中电泳时,由于琼脂糖凝胶具有一定孔径,长度不同的DNA分子由于所受凝胶的阻遏作用大小不一,迁移的速度不同,从而可以按照分子量大小得到有效分离。
溴化乙锭(EB)可插入到DNA分子的相邻碱基之间,在紫外光的照射下出现荧光,从而指示DNA含量和位置。
实验仪器:
电泳仪、紫外检测仪、水平电泳槽、移液枪、一次性手套
TAE、EB溶液、凝胶加样缓冲液、琼脂糖
1、1%琼脂糖凝胶的制备(以制备5块胶为例):
(1)称取琼脂糖1.6g,加入160ml1xTAE缓冲液,在微波炉上加热溶解煮沸熔化,取出摇匀至无沉淀。
(2)取有机玻璃内槽,放置于水平位置,用移液枪吸取少量胶将有机玻璃内槽两端密封。
冷却到60℃左右(手感温度不烫手即可)加入7ulEB溶液,摇匀后倒入有机玻璃内槽,等距插入5个样品梳子。
(3)冷凝后将梳子拨出,电泳胶放入电泳槽中,加入TAE电泳缓冲液没过胶面。
2、加样
用移液枪取缓冲液,依次点3ul缓冲液在点样板上,再取PCR样品溶液10ul左右与缓冲液混匀后,加入样品孔中,并记录好点样顺序。
(大体系PCR产物需要回收,点样时更换枪头避免交叉污染)
3、电泳及结果观察
接通电泳槽与电泳仪的电源,170V恒压电泳20min,电泳至溴酚蓝接近胶底部边缘即可;
电泳完毕,取出凝胶,放在紫外灯下观察DNA条带的位置,根据DNA条带与溴酚蓝的相对位置估算DNA分子量大小,记录结果。
三、DNA片段回收:
1、摇匀GS树脂,用移液枪取500ul树脂分装于1.5mlEP管中,将PCR大体系产物加入到GS树脂中,用移液枪吹打混匀,静置10min,让片段结合到GS树脂上
2、将混合液转入纯化柱中,13000rpm瞬时离心,弃收集管中液体
3、加600ul80%异丙醇洗脱两次,13000rpm瞬时离心,弃收集管中液体,13000rpm空甩20min;
取出回收柱放入干净的1.5mlEP管中,放入30-40摄氏度烘箱烘干10min左右,使异丙醇完全挥发。
4、取出加入50ulddH2O浸润树脂干粉,静置10min让ddH2O充分浸润树脂干粉,13000rpm离心2-3min,得到的PCR产物用于双酶切。
实验结果:
序号
引物号
20ul小体系
片段大小
1
P18279
√
327
P18280
339
3
P18281
378
4
P18282
321
5
P18283
552
6
P18284
231
7
P18285
222
8
P18286
×
9
P18287
501
P18288
528
11
P18290
471
12
P18291
351
13
P18292
186
14
P19293
645
15
P18295
873
16
P18296
612
17
P18297
243
18
P18298
417
19
P18299
903
20
P18300
249
21
P18302
285
22
P18303
300
23
P18304
306
24
P18305
345
25
P18289
26
P18274
489
实验二DNA双酶切及酶切产物纯化
通过DNA限制性内切酶对DNA双链进行双酶切,使DNA片段露出相应的粘性末端。
1、核酸限制性内切酶是在原核生物中发现的一类专一识别双链DNA中特定碱基序列的核酸水解酶,他们的功能类似动物的免疫系统,用于抗击外来DNA的侵袭,具有自我保护机制。
一种限制性内切酶只能识别一种特定的核苷酸序列,并在特定的位点上以内切方式水解核酸链中的磷酸二酯键,切割DNA分子。
在酶切反应中,DNA的纯度、缓冲液中的离子强度、Mg2+等因素均可影响酶切反应,一般可通过增加酶的用量,延长反应时间等措施以达到完全酶切。
2、Ⅱ型限制性核酸内切酶是基因工程中剪切DNA分子的常用工具酶,其优点是:
只具有限制性活性,无甲基化修饰活性;
对DNA的切割要求严格的序列作为靶位点,切割精确;
此类酶作用时只需要Mg2+参与,无需ATP。
3、DNA双酶切可有效防止载体自连形成空载体,双酶切应选择公用的Buffer
PCR仪、PCR管、移液枪、tip枪头、限制性内切酶
取5ul通用酶切bufferTango,加入1ul限制性内切酶1和1ul限制性内切酶2,将PCR回收产物全部(大约43ul)加入混匀,体系配好后盖上PCR管并做好编号,放入PCR仪中,设定37℃酶切过夜。
用含有GS结合液的树脂纯化酶切产物,操作方法同PCR产物回收方法。
酶切位点1
酶切位点2
Buffer
EcoRⅠ
XholⅠ
Tango
BamhⅠ
EcorⅠ
P18293
实验三目的基因与载体连接
将外源DNA片段与线状质粒载体连接,构建具有目的基因的质粒载体。
在Mg2+和ATP存在下,DNA连接酶能催化载体分子的粘性末端与外源DNA的相同粘性末端连接成重组DNA分子。
T4DNA连接酶与辅助因子ATP形成酶-AMP复合物;
酶-AMP复合物结合到相邻的具有5'
磷酸和3'
羟基切口的DNA上,使DNA腺苷化,形成一个新的磷酸二酯键,将切口封闭。
实验仪器与材料:
PCR仪、PCR管、移液枪、tip枪头、T4连接酶Buffer、T4DNA连接酶、pet-28a载体、pGEX-4T载体
实验步骤
1、配制连接反应体系,按顺序向微量PCR管中依次加入:
10×
T4连接酶buffer1ul
T4连接酶1ul
PEGX载体/PET28a载体1ul
纯化后酶切产物7ul
2、体系配好后放置于PCR仪中,22℃保温4h。
第一批连接重组22个
PGEX-4TE+X
18279、18292、18299
PGEX-4TB+X
18280、18281、18282、18383、18284、18285、18287、18290
pet28aE+X
pet28aB+X
第二批连接重组24个
18291、18293、18296、18297、18298、18300、18302、18303、18304、18305、18289、18274
实验四重组子转化
试验目的:
将含有目的基因的重组质粒转化入大肠杆菌中。
试验原理:
细菌处于容易吸收外源DNA的状态叫做感受态。
细菌在0℃CaCl2低渗溶液中胀成球形,丢失部分膜蛋白,成为容易吸收外源DNA的状态。
细菌处于0℃CaCl2低渗溶液中,外源DNA形成抗DNA酶的羟基-钙磷酸盐复合物粘附于细胞表面,经过42°
C90S热击处理,促进吸收DNA复合物。
然后在非选择培养基中培养一代,待质粒上所带的抗药性基因表达,即可在含Amp或Kan的培养基中生长。
从而筛选出重组子,即带有外源DNA分子的受体细胞。
超净工作台、低温离心机、恒温摇床、恒温箱、-80℃冰箱、恒温水浴锅
胰蛋白胨、酵母提取物、氯化钠(Nacl)、氨苄青霉素、感受态DE3(BL21)、pet-28a载体、pGEX-4T载体、1.5mL离心管、移液枪、试管、培养皿等
试剂准备:
(1)SOB培养基200ml:
蛋白胨4g、酵母粉1g、NaCl0.1g,加水定容至200ml,调节pH7.0
(2)LB固体培养基:
1%NaCl、1%胰蛋白胨、0.5%酵母提取物、1.5%琼脂粉,培养基放入121℃湿热灭菌20min。
(3)抗生素:
卡那青霉素,氨苄青霉素
1、连接完成的重组质粒,在PCR仪中65℃灭活10min
2、从-80℃冰箱中取2支感受态细胞,在酒精灯火焰下,分装80ul感受态于1.5mlEP管中,并加入10ul重组质粒混匀,冰浴30分钟。
3、于42℃恒温水浴锅中热激90s,迅速转移至冰浴中,继续冰浴5min
4、每管加入800ulSOB、16ulGlu、4ulMgCl2,37℃150rpm摇床培养50min。
5、8000rpm离心5min,倒掉部分上清,在超净工作台中将吹打混匀后培养物涂布于含Amp或Kan的LB琼脂平板上。
6、在37℃培养箱中将平皿倒置,培养过夜12-16h。
在含氨苄青霉素的培养基上生长的菌落即为含有pGEX-4T重组载体的大肠杆菌,在含卡那青霉素的培养基上生长的菌落即为含有pet-28a重组载体的大肠杆菌。
两批实验连接的18304构建Amp和Kan抗性平板上均未长,可能是酶切错误导致。
后重新PCR、酶切重连,转化筛选呈阳性,但未表达。
实验五重组子筛选和接种
从单抗菌落中筛选出阳性克隆
带有目的基因的单克隆菌落在,在高温下(95℃)裂解释放出目的基因的质粒,通过PCR扩增目的基因,从而达到检测的目的。
恒温培养箱、PCR仪、电泳仪、紫外检测仪、水平电泳槽、移液枪、一次性手套、LB固体培养基、牙签,PCR扩增试剂等
1、取相应抗性的平板,在平板上画格子;
配制PCR反应体系
2、取出过夜培养的平板,每个菌筛选5个单克隆菌落在平板格子内画线,画线的牙签放入配好的20ul体系中涮洗,涮洗过牙签的体系放入PCR仪中进行扩增,画线的平板放在37℃培养箱中倒置培养4h。
3、PCR完成后,1%琼脂糖凝胶电泳检测筛选结果。
第一批筛选结果记录
氨苄青霉素抗性
卡那青霉素抗性
3、4、5
1、2、4
1、2、3、4、5
1、4、5
-
1、3、4、5
2、3、4、5
1、2、3、5
1、2、3、4
1、2、5
1、2、4、5
1、3
4、5
第二批筛选结果记录
1、3、
2、4
1、2
3、4
2、3
2、3、4
平板未长
小样接种
挑取两个阳性克隆菌落,接种于4ml液体LB抗性培养基中,37℃220rpm摇床过夜培养,两批接种信息如下:
名称
+18279-3/-4
+K18279-1/-2
+18291-1/-3
+K18291-1/-3
+18280-1/-2
+K18280-1/-4
+18293-1/-2
+K18293-2/-4
+18281-1/-4
+K18281-1/-2
+18296-1/-2
+K18296-2/-3
+18282-3/-4
+K18282-1/-2
+18297-3/-4
+K18297-1/-2
+18283-1/-3
+K18283-1/-2
+18298-4/-5
+K18298-2/-3
+18284-1/-2
+K18284-2/-3
+18300-3/-4
+K18300-1/-2
+18285-1/-2
+K18285-1/-2
+18302-2/-3
+K18302-2/-3
+18287-1/-2
+K18287-1/-2
+18303-2/-4
+K18303-1/-2
+18290-1/-3
+K18290-2/-3
+18305-2/-3
+K18305-2/-3
+18299-1/-2
+K18299-1/-3
+18289-1/-2
+K18289-3/-4
+18292-1/-2
+K18292-4/-5
+18274-1/-2
+K18274-1/-2
小样接种菌生长良好,无未长现象。
实验六外源基因在大肠杆菌中的诱导表达与制样
检测阳性克隆的表达情况
1、EcoliLac操纵子包括Z、Y、A三个结构基因,以及启动子P、控制子O、阻遏子I。
LacI基因编码一种阻遏蛋白,其与操纵序列O结合,使基因表达处于关闭状态。
2、在没有乳糖存在时,Lac操纵子处于阻遏状态,此时I序列在启动子的作用下表达阻遏蛋白,并与O序列结合,抑制转录的起始;
当有乳糖存在时,Lac操纵子即可被诱导,真正的诱导剂并非乳糖本身,乳糖进入细胞后,在β-半乳糖苷酶作用下转变为半乳糖,后者作为一种诱导剂结合阻遏蛋白,从而使RNA聚合酶能顺利地与控制子结合并移动,使LacZ、Y、A基因得以表达。
3、异丙基巯基半乳糖苷(IPTG),是一种乳糖类似物,因为不是半乳糖苷酶的底物,所以不能被细胞利用,从而实现持续表达。
恒温摇床、玻璃试管、超净工作台、低温离心机、湿热灭菌锅、含外源基因表达栽体的E.coli、酵母粉、胰化蛋白胨(tryptone)、氯化钠(Nacl)、甘油、氨苄青霉素、卡那青霉素、IPTG、移液枪、tip枪头
1、取200ul培养过夜的菌种,接种于新的4mLLB抗生素培养基中
2、于37℃恒温摇床,200r/min培养1.5h后,加入30μlIPTG,继续培养3.5小时。
3、取1.5ml菌液,8000rpm,低温离心5min,收集菌体弃上清,加入SDSBuffer100ul,用牙签搅碎菌体,放在沸水中煮沸1.5-2h,于4°
C冰箱中保存。
扩大接种和诱导培养,无变清现象,菌体生长良好。
实验七Western-blot检测表达蛋白
蛋白经SDS-PAGE后进行Western-blot检测,鉴定目的蛋白的表达情况
1、SDS-PAGE电泳分离蛋白质,是基于各蛋白组分在凝胶中的迁移率主要取决于分子大小和形状,及其所带电荷的多少。
在聚丙烯酰胺凝胶系统中,加入一定量的SDS,SDS与蛋白分子结合,使其带上相同密度的负电荷,其数量远远超过蛋白分子原有的电荷量,从而掩盖了不同蛋白质间原有的电荷差异,因而蛋白质分子的电泳迁移率主要取决于分子量大小。
2、WesternBlot法采用的是聚丙烯酰胺凝胶电泳检测蛋白质,利用抗体作为“探针”,用二抗标记“显色”。
经过SDS-PAGE分离的蛋白质样品,转移到固相载体(例如硝酸纤维素薄膜)上,固相载体以非共价键形式吸附蛋白质,且能保持电泳分离的多肽类型及其生物学活性不变。
以固相载体上的蛋白质或多肽作为抗原,与对应的抗体起免疫反应,再与酶或同位素标记的第二抗体起反应,经过底物显色或放射自显影以检测目的基因表达的蛋白成分。
一、SDS-PAGE电泳
1.配制分离胶
配制12%和14%分离胶按顺序和量(注意体积单位!
)取下列溶液混在一个50ml的小烧杯中:
凝胶浓度(%)
分离胶buffer
4ml
40%gel
2ml
2.2ml
双蒸水
1.8ml
TEMED
8ul
10%Aps
80ul
总体积
8ml
不同浓度的凝胶的成分和用量
根据被分离的外源蛋白的相对分子量选择凝胶浓度,先洗净玻璃架好制胶架,装满水静置一段时间,不漏水为宜;
将所有成分混匀后加入两玻璃夹缝中,胶面离小玻璃上端1cm左右,并小心在胶面上加入1cm蒸馏水,约30min等胶自然凝聚后倾斜倒出蒸馏水沥干。
2.4%浓缩胶的配置按顺序和量(注意体积单位!
4%分离胶的配制成分
成分
用量
压缩胶buffer
1.5ml
0.3ml
1.2ml
4ul
40ul
3ml
混匀后,立刻转移到玻璃夹缝中,液面与玻璃上边缘齐平,并在两玻璃夹缝中快速斜插入15孔的梳子,溶液凝固后小心拔出梳子。
3.上样氨苄抗性加10ul,卡那抗性加15ul,marker加样量随样。
4.电泳接好电极,将电压调至80V,待溴酚蓝移到分离胶后(约20min),在将电压调至120V,当溴酚蓝移到玻璃距底部即将全部跑出时切断电源,总用时大约90min。
二、转膜
(1)将上层滤纸、下层滤纸分开浸泡于transferbuffer中,用镊子将编号的PVDF膜放在甲醇中浸润2-3min后浸泡在transferbuffer中。
(2)在半干转移仪阳极面铺好下层滤纸;
将膜铺在下层滤纸上,不要产生气泡,倒少量Buffer于膜上,保持膜的湿润;
将胶取出,切掉上层胶,小心将胶铺在膜上,赶走气泡;
将上层滤纸覆盖在胶上,按住一端,用滚轮赶走多余的液体。
(注意滤纸与膜、膜与胶之间不能有气泡)
(3)盖上电极盖子,接通电源,每张膜恒流60mA,转移40min。
三、蛋白印迹
1、封闭位点转移结束前配好5%TBST牛奶,转移完毕后将膜放入甲醇浸泡,然后晾干使膜完全疏水
2、加一抗孵育每个盒子放两张膜,背面相贴而放,加30ml5%TBST牛奶封闭位点,按1:
8000加GST、His抗体孵育1h
3、洗涤孵育完后,用TBST快洗三次,再在转摇床洗5次,每次5min,洗掉牛奶。
4、DAB显色取一管DAB加入15ul过氧化氢,将洗涤后的膜放入染色10min左右
5、用自来水清洗膜数次后,晾干观察。
第一批连接Western-blot结果图
18279-4K18279-1
18280-1K18280-1
18282-3K18282-1
18283-1K18283-1
18284-1K18284-2
18285-1K18287


- 配套讲稿:
如PPT文件的首页显示word图标,表示该PPT已包含配套word讲稿。双击word图标可打开word文档。
- 特殊限制:
部分文档作品中含有的国旗、国徽等图片,仅作为作品整体效果示例展示,禁止商用。设计者仅对作品中独创性部分享有著作权。
- 关 键 词:
- 融合 蛋白 表达 实验 报告
 冰豆网所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
冰豆网所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
 铝散热器项目年度预算报告.docx
铝散热器项目年度预算报告.docx
-
牛津上海版通用小学英语三年级上册Unit 12同步练习2II 卷.docx
-
论我国私营企业员工激励机制.docx
-
人教版五年级品德与社会上册全册教案.docx
-
开学啦国旗下讲话稿三分钟.docx
-
露天采矿学复习题.docx
-
六年级英语教师年度考核个人总结.docx
-
某路站综合体项PC吊装施工方案.docx
-
人教版九年级历史上册期末考试试题一套.docx
-
隆昌妇幼保健院.docx
-
芦二矿抽采达标中长期规划.docx
-
看拼音写词语.docx
-
模拟磁盘调度算法系统的设计毕业设计.docx
-
每周一条名言警句或一首诗词.docx
-
棉花膜下滴灌示范工程设计总结报告.docx
-
九年级化学教案第十单元酸和碱教案新人教版.docx
-
宁波市水资源公报.docx
-
农业实用技术培训工作意见与农业局上半年工作总结范例两篇汇编.docx
-
平行线的判定.docx
-
内部会计管理制度11成本核算制度.docx
-
盘扣式脚手架支撑方案.docx
-
旅游规划模板.docx
-
煤矿大本大专毕业设计大采高综采工作面作业规程.docx
-
美学选择题整理课件资料.docx
-
名家论腹泻慢性肠炎.docx
-
宁夏银川市第一中学学年高一上学期期中考试地理试题解析解析版.docx
-
年产吨精密纤维纸项目建设建议书.docx
-
农技推广中心工作总结.docx
-
彭宇案的法逻辑批判.docx
-
宁夏仕奇房产网发布份房地产交易情况.docx
-
项目推荐书智能温控节能系统.docx
-
区县节日期间加强消防安全讲话稿与区发改委领导班子述职述廉报告汇编.docx
-
东方红表演主持词.docx
-
大学毕业祝福语简短励志大学毕业祝福语创意.docx
-
冬季雨季施工方案.docx
-
社区治理.docx
-
大学生贫困补助申请书怎么写.docx
-
短语结构类型.docx
-
动物遗传学试题库及答案.docx
-
大学生职业生涯规划书87814.docx
-
读书社团活动计划3篇.docx
-
多篇银行职业发展规划可参考.docx
-
申论常见错误.docx
-
砂夹石回填施工方案 已修改.docx
-
儿童节小笑话.docx
-
山东省17市三年中考化学试题分类汇编气体制备含答案.docx
-
对中小企业财务管理问题的思考.docx
-
导学单备课模板.docx
-
二次函数的图像与性质专题练习.docx
-
山东省临沂市届高三份教学质量检测语文试题 Word版含答案.docx
-
生活更美好作文500字15篇.docx
